
技术摘要:
本发明公开了一种新型二氢吡咯并[1,2‑a]吲哚衍生物及其合成方法。具体合成方法是将反应物A、反应物B、催化剂以及碱于有机溶剂中发生反应,经过分离提纯,得到所述二氢吡咯并[1,2‑a]吲哚衍生物。本发明的合成方法具有反应区域选择性、反应条件温和、反应及后处理纯化 全部
背景技术:
稠环[1,2-a]吲哚类衍生物广泛存在于自然界中,是很多具有重要生物活性和药 用价值的天然产物以及药物分子的核心骨架,其中二氢吡咯并吲哚(结构如附图1所示)作 为重要的稠三环吲哚骨架分子,具有良好的药理活性。例如,二氢吡咯并吲哚类化合物对蛋 白酪氨酸磷酸酶有较好的抑制作用,在糖尿病和肥胖症的治疗中具有潜在的应用价值。传 统的从自然界中提取吲哚类化合物已难以满足现代工业需求,因此为这类稠三环吲哚类化 合物提供高效、简便的合成方法一直是现代有机合成工作者的研究热点。
技术实现要素:
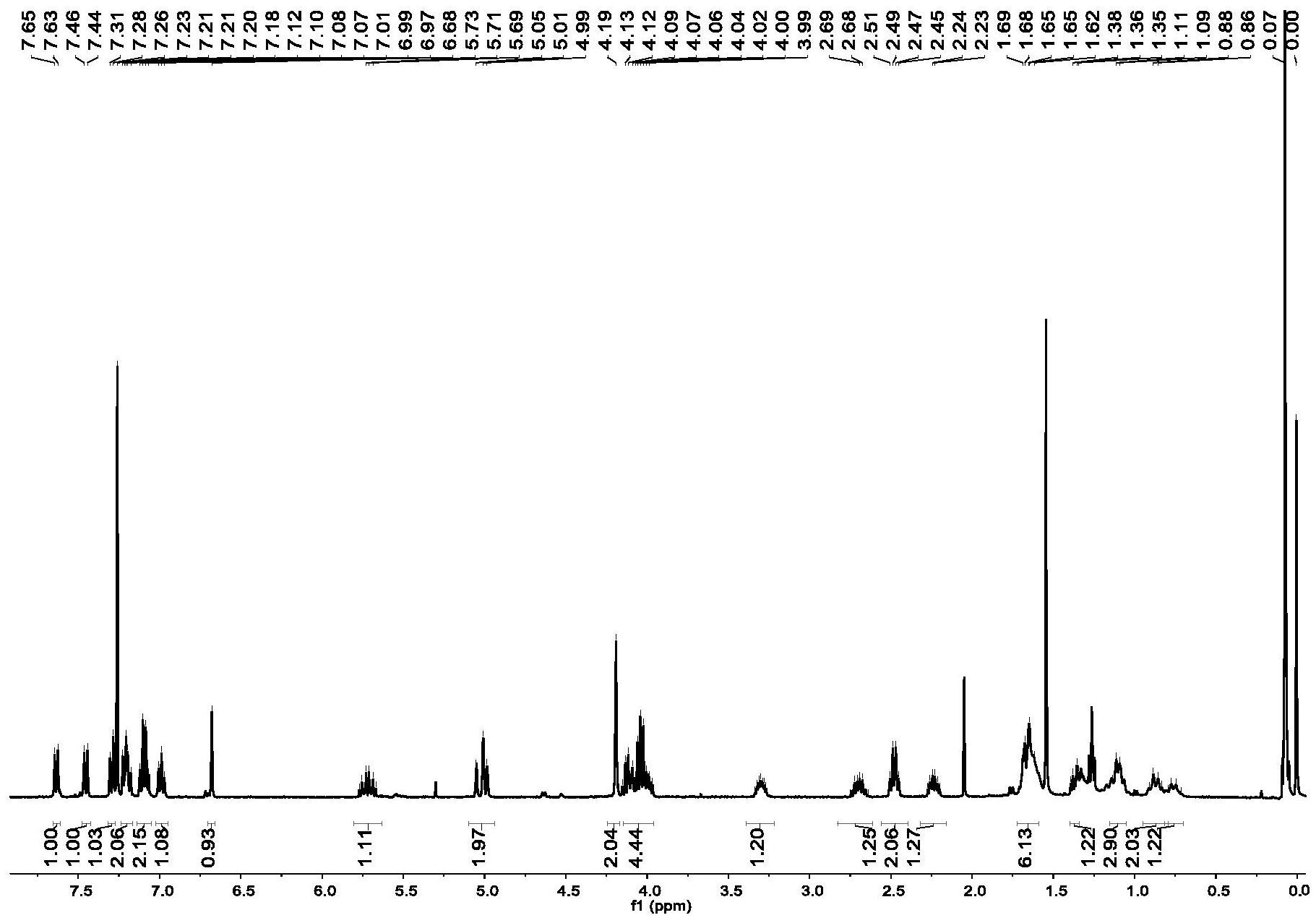
本发明通过吲哚化合物的环化反应,设计以吲哚作为控制基团来控制反应中的区 域和化学选择性,成功合成了一种新型的二氢吡咯并[1,2-a]吲哚衍生物,该合成方法具有 反应区域选择性、反应条件温和、反应及后处理纯化过程简单的优点。 为了解决上述技术问题,本发明提供了如下的技术方案: 本发明提供第一方面提供了一种二氢吡咯并[1,2-a]吲哚衍生物,所述衍生物具 有如下所示的结构式: 其中,R1为H、甲基、乙基、乙酰基或羧甲基; R2为: 本发明第二方面提供了如第一方面所述的二氢吡咯并[1,2-a]吲哚衍生物的合成 方法,包括以下步骤: 在惰性气氛下,将反应物A、反应物B、催化剂以及碱于有机溶剂中发生反应,经过 分离提纯,得到所述二氢吡咯并[1,2-a]吲哚衍生物; 所述反应物A的结构式为 R1为H、甲基、乙基、乙酰基或羧甲 基; 4 CN 111592545 A 说 明 书 2/3 页 所述反应物B的结构式为 R2为 所述催化剂是由靶催化剂与双膦配体组成的络合物; 所述碱为氮甲基有机胺; 所述有机溶剂为芳香族有机溶剂。 上述反应的过程如下所示: 进一步地,所述靶催化剂包括Pd(pph3)4、Pd(OAc)2、Pd(OTf)2、Pd(TFA)2、PdCl2、 Na2PdCl4、PdSO4、Pd(dba)2、Pd2(dba)3,优选为Pd(pph3)4。 进一步地,所述双膦配体包括PhPCy2、t-Bu3P、PCy3、dppe、dppp、dppb、dppf,优选为 dppp。 进一步地,所述碱为N-甲基二环己基胺、二乙胺、三乙胺或质子碱,优选为N-甲基 二环己基胺。 进一步地,所述有机溶剂选自苯、甲苯、二甲苯、三氟甲苯中的一种或多种的组合, 优选为无水三氟甲苯。 进一步地,所述反应物A、反应物B、催化剂以及碱的摩尔比为(2.5~5):(1~2): (0.1~0.5):(3~5)。 进一步地,所述反应物A为N-丁烯吲哚,反应物B为碘代环己烷,钯催化剂为四(三 苯基膦)钯,双膦配体为1,3-双(二苯基膦)丙烷,碱为N-甲基二环己基胺,有机溶剂为无水 三氟甲苯。 进一步地,各组分的摩尔比为: 碘代环己烷:N-丁烯吲哚:四(三苯基膦)钯:1,3-双(二苯基膦)丙烷:N-甲基二环 己基胺=1∶2.5∶0.1∶0.15∶3。 进一步地,所述反应温度为110~130℃,反应时间为24~48h。优选地,反应于油浴 中剧烈搅拌下进行,油浴温度优选为110℃,反应时间优选为24h。 进一步地,反应结束后,产物通过硅胶柱纯化。 与现有技术相比,本发明的有益效果在于: 1.本发明通过设计以吲哚类芳香骨架作为控制基团来调控环化反应,使烷基选择 性地加成到双键上,成功合成了一种新型二氢吡咯并[1,2-a]吲哚衍生物。本发明的合成方 5 CN 111592545 A 说 明 书 3/3 页 法,通过一步环化反应即可构建复杂的多核心吲哚衍生物,反应过程简单;且该合成方法具 有反应区域选择性、反应条件温和、后处理纯化过程简单等优点。 2.本发明合成的二氢吡咯并[1,2-a]吲哚衍生物,可作为药物合成的中间体,具有 广泛的应用前景。 附图说明 图1为二氢吡咯并吲哚的结构式; 图2为具有二氢吡咯并吲哚结构的天然生物活性分子; 图3是实施例1合成的二氢吡咯并[1,2-a]吲哚衍生物的核磁氢谱图。











