
技术摘要:
本发明涉及一种机器学习模型高通量分析乙型肝炎病毒基因组RT/S区序列特征预测肝癌风险的方法,属于基因组序列计算技术领域。本发明选用相应的特征计算方法完成HBV基因组RT/S区序列特征矩阵的构建;再采用机器学习模型对数据集进行训练,最后进行是否罹患肝癌风险概率预 全部
背景技术:
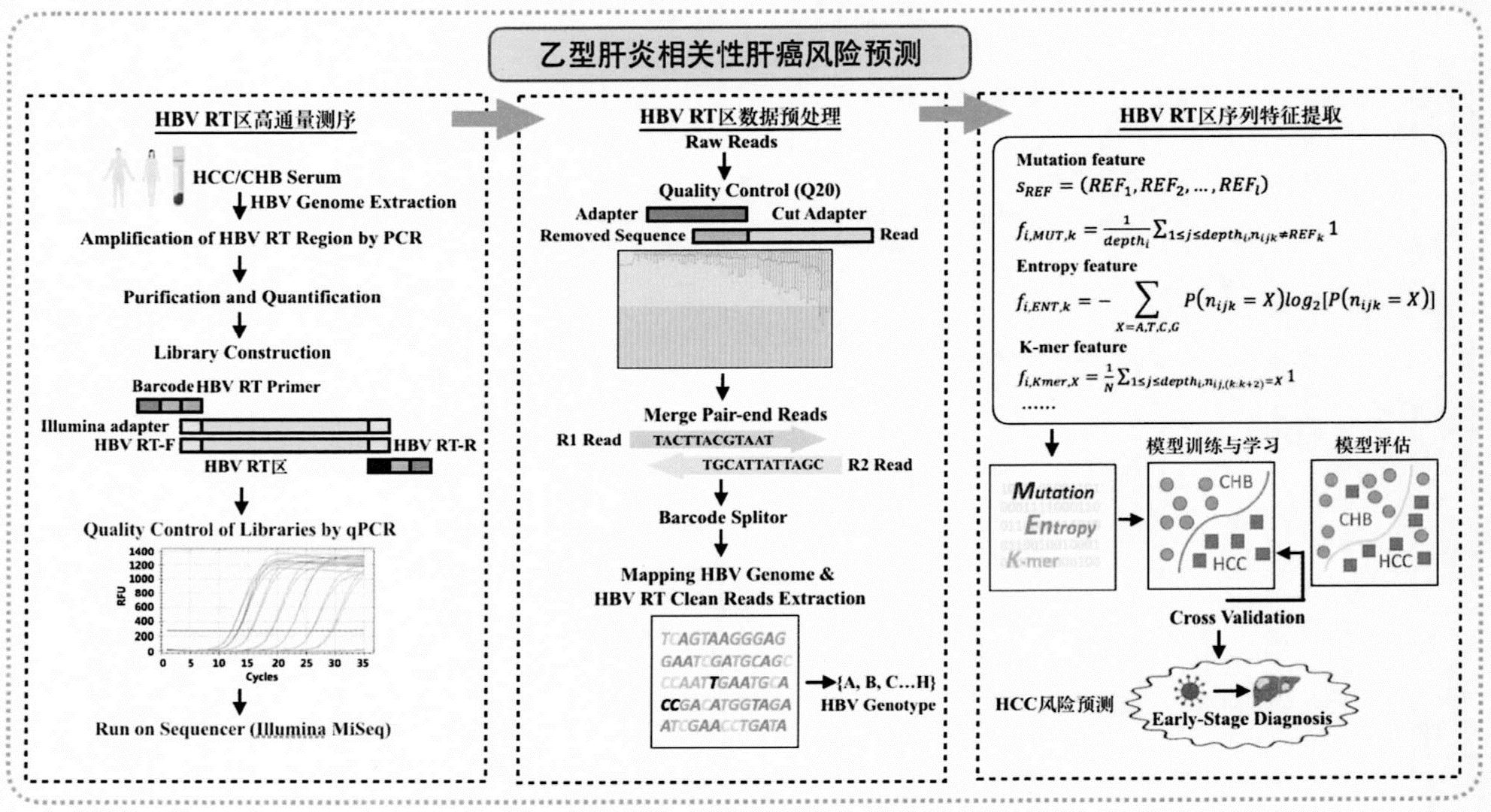
肝细胞癌(Hepatocellular carcinoma,HCC)是最常见的原发性肝癌,是癌症死亡 的第三大主要原因,也是世界上第六大最常见的癌症。HBV感染是最重要的危险因素之一。 据世界卫生组织(WHO)估计,全球约有2.57亿人被确认为乙肝病毒表面抗原(Hepatitis B virus surface antigen,HBsAg)阳性。慢性乙肝患者(Chronic hepatitis B,CHB)若不及 时治疗干预,将逐渐发展为肝硬化甚至肝癌。 HBV基因组是一个有部分单链区的环状双链DNA分子,由四个重叠的开放阅读框 (ORF):P、C、S 和X区组成。P-ORF中的HBV逆转录酶(RT)基因编码聚合酶蛋白的RT区域,其中 有部分与S基因编码HBsAg重叠,是抗病毒耐药性和免疫逃逸的重要区域。HBV基因组的突变 与HCC密切相关,主要是由于HBV的高复制率高,宿主免疫压力和易于出错的HBV逆转录酶。 目前,大多数研究都基于Sanger测序检测HBV基因变异。然而,由于灵敏度低(约 20%),传统的 Sanger测序难以识别较小的病毒变异并难以表征单个患者体内HBV准种的 复杂模式。与一代测序相对比,二代测序技术(Next generation sequencing,NGS)能大量 获取HBV准种序列片段的同时,保持高灵敏度和高特异性。它能够精准而深入的探索HBV突 变信息,并且基于大量序列信息进一步取序列特征再进行生物信息学分析。然而,迄今为止 很少有研究通过使用NGS技术产生的大数据对CHB和HBV相关的HCC 患者进行HBV RT基因突 变特征的分析与研究。此外,传统的统计算法似乎不足以处理此类高维基因组数据,无法将 质量测序信号转化为可用于临床的可行生物标记。 针对上述问题,本发明对NGS测序产生的HBV基因组RT/S区序列数据进行特征计 算,并整合机器学习模型,以鉴定HBVRT/S区的内部准种特征,进行个体HCC风险预测。该方 法用于HCC风险筛查、辅助临床诊断和疾病分层管理方面具有重要临床应用前景,是第一个 利用动态HBV准种特征和机器学习模型进行HCC风险预测的计算方法。
技术实现要素:
本发明要解决的技术问题是针对现有NGS技术在处理高维HBV基因组数据和有效 转化为可用于临床的肝癌风险模型方面的不足,提供一种面向机器学习模型的HBV基因组 RT/S区序列特征预测肝癌风险的方法,将算法生成的特征矩阵应用于基因组序列数据处理 的机器学习模型训练学习,通过建立分类模型,实现对CHB(非HCC)和HCC人群的鉴别,以期 其可在HCC的辅助诊断及临床高危预警中发挥作用。 3 CN 111613324 A 说 明 书 2/6 页 为解决上述技术问题,本发明提供一种采用机器学习模型高通量分析HBV基因组 RT/S区序列特征预测肝癌风险的方法,包括以下步骤: (1) .数据预处理:对于序列数据,判断序列存储类型是否为FASTA格式,判断是否 为反向互补序列,并结合判断结果,选用相应的方法进行预处理; (2) .生成RT区及S区氨基酸序列; (3) .对于RT区核苷酸序列及RT区/S区氨基酸序列分别进行特征矩阵构建; (4) .将特征矩阵数据集划分为训练集与测试集,将训练集输入机器学习模型中进 行参数训练,并进行概率预测。 (5) .使用训练好的模型,针对新的一批患者数据,评估模型的鲁棒性和各类性能, 如敏感性、特异性、 AUC值、准确率等。 所述步骤(2)中,RT区氨基酸序列的翻译从第1位核苷酸起始,S区氨基酸序列的翻 译从第2位核苷酸起始,采用参考序列(https://www .ncbi .nlm .nih .gov/projects/ genotyping/view.cgi?db=2)。 所述步骤(3)中,特征算法为位点的碱基分布百分比特征(ACGT feature)、RT区核 苷酸序列突变频率特征(Mutation feature)、RT区氨基酸序列突变频率特征(Mutation feature of HBV RT amino acid)、S区氨基酸序列突变频率特征(Mutation feature of HBsAg amino acid)、RT区核苷酸序列香农熵特征(Entropy feature)、RT区氨基酸序列香 农熵特征(Entropy feature of HBV rt amino acid)、S区氨基酸序列香农熵特征 (Entropy feature of HBsAg amino acid)、RT区核苷酸序列游走香农熵特征(Group Walk entropy feature)、 RT区核苷酸序列滑动香农熵特征(Slide entropy feature)和RT区序 列3-mer特征(K-mer feature)十种。 所述步骤(4)中,机器学习训练模型为随机森林(Random Forests,RF)、K最近邻 (k-Nearest Neighbor, kNN)、支持向量机(Support Vector Machine,SVM)和逻辑回归 (Logistic Regression,LR)四种。采用的机器学习模型都有较好的适用性。较为典型的是, 在分类模型RF中,针对不同HBV基因型和测序深度的分类能有效提升预测准确率。 本方法中,针对HBV基因组数据RT/S区序列分为两个片段,RT1序列长度为481bp, RT2序列长度为406bp。 本方法中,对于每一个训练的模型,共输入了11种不同的特征(10种单一特征以及 1种所有特征的组合),最终一共得到44种不同模型。 所述方法的特征矩阵还加入了all features特征,包含十种特征的整合,以提高 模型的预测能力。在模型训练与测试完成后,选择曲线下面积AUC最优的模型作为CHB和HCC 人群进行鉴别的模型,模型输出值作为HCC风险预测得分。所建立的模型经独立样本验证同 样具有很好的曲线下面积。 本发明基于人工智能的机器学习模型,将序列特征算法应用在NGS测序数据处理 领域的机器学习模型训练上,可以有效地解决NGS高维序列数据的处理与临床应用,有助于 利用机器学习的方式进行计算机辅助筛查HCC高危人群,提高对该病的检出率和临床精准 治疗。 4 CN 111613324 A 说 明 书 3/6 页 附图说明 图1是本发明实施例采用机器学习方法训练与预测的样本分类AUC评估图。 图2是本发明实施例用于两种HBV基因型人群的模型分类性能对比图。 图3是本发明实施例用于不同测序深度的模型分类性能对比图。 图4是本发明乙型肝炎相关性肝癌风险预测的流程图。











