
技术摘要:
本发明涉及药物质量检测技术领域,特别涉及检测头孢克肟聚合物杂质的方法。该方法包括使用高效液相色谱法检测,所述高效液相色谱法的检测条件包括:流动相:由流动相A和流动相B组成,其中,流动相A为甲酸溶液,流动相B为乙腈、甲醇或乙腈‑甲醇;流动相A和流动相B的体 全部
背景技术:
头孢克肟(Cefixime)属于β-内酰胺类抗生素,是第三代头孢菌素类抗生素,其化 学名称为:(6R,7R)-7-[(Z)-2-(2-氨基-4-噻唑基)-2-(羧甲氧亚氨基)乙酰氨基]-8-氧代- 3-乙烯-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸三水合物;具有半衰期长、高效低毒、 体内分布广、组织穿透性好等特点。 头孢菌素类抗生素中的聚合物杂质的来源可分为两类:外源性杂质和内源性杂 质。外源性杂质一般来源于发酵工艺,为蛋白、多肽、多糖等杂质与抗生素结合的杂质;内源 性杂质系指抗生素药物自身聚合产物,聚合物既可来自生产过程,又可在储存过程中形成。 头孢菌素的聚合反应包括只依赖于母核的N型聚合反应和侧链参与的L型聚合反应,固体状 态下头孢菌素类产品的聚合速度与含水量和贮存温度关系密切。目前,对于头孢菌素类抗 生素中的聚合物杂质的检测多采用凝胶色谱法进行检测分析,凝胶色谱法又称为分子排阻 色谱法,主要是利用分子筛机制,分子质量较小的药物分子进入凝胶孔隙内部,保留时间较 长,而分子质量较大的聚合物杂质被排阻在凝胶孔隙之外,保留时间较短,较早被洗脱,从 而与药物分子分离。对于头孢克肟原料药和制剂中聚合物杂质的测定,各国药典均无记载。 现有技术中,赵晓东等学者(赵晓冬,傅蓉,陆军,等.分子排阻色谱法测定头孢克肟聚合物 [J].中国药师,2011 ,14(1):61-63.)建立了分子排阻法分析头孢克肟高分子聚合物的方 法,采用SephadexG10色谱柱(300mm×16mm,40~120μm),流动相A为0.1mol/L磷酸缓冲液 (pH7.0),流动相B为超纯水,流速1.5mL/min,检测波长为254nm。结果显示,头孢克肟在5~ 80mg/mL的范围内与头孢克肟高分子聚合物的峰面积呈良好的线性关系(r=0.9993),头孢 克肟在5~400μg/mL的范围内与头孢克肟缔合物的峰面积也呈良好的线性关系(r= 0.9997)。然而,从色谱图上可以明显看出,头孢克肟高聚物的峰型较差。申请人在早期也对 头孢克肟聚合物杂质的检测方法进行了研究,采用传统的凝胶色谱法,所用色谱柱为TSK gel G2000 SWXL 7.8mm*30cm*5μm,色谱条件:HPLC-UV,波长:254nm,流速:0.8mL/min,流动 相:pH7.0 0.005mol/L磷酸盐缓冲液[0.005mol/磷酸氢二钠溶液-0.005mol/L磷酸二氢钠 溶液(61:39)]-乙腈(95:5),进样量:10μL,供试品浓度:0.5mg/ml,稀释液:水,对照或标准 溶液:10μg/ml,计算方法:外标法,运行时间:20min,杂质限度控制:总杂≤1.0%。通过分段 收集高比例有机相梯度洗脱方法分离的含不同杂质的洗脱液,在凝胶色谱柱法中进行定 位,发现在主成分头孢克肟出峰前洗脱出来的理论上为高分子杂质,即头孢克肟聚合物的 杂质,但绝大部分分子量与头孢克肟接近。表明凝胶色谱法测定头孢克肟聚合物的专属差。 可见,对于头孢克肟聚合物的测定方法需要重新探索开发。
技术实现要素:
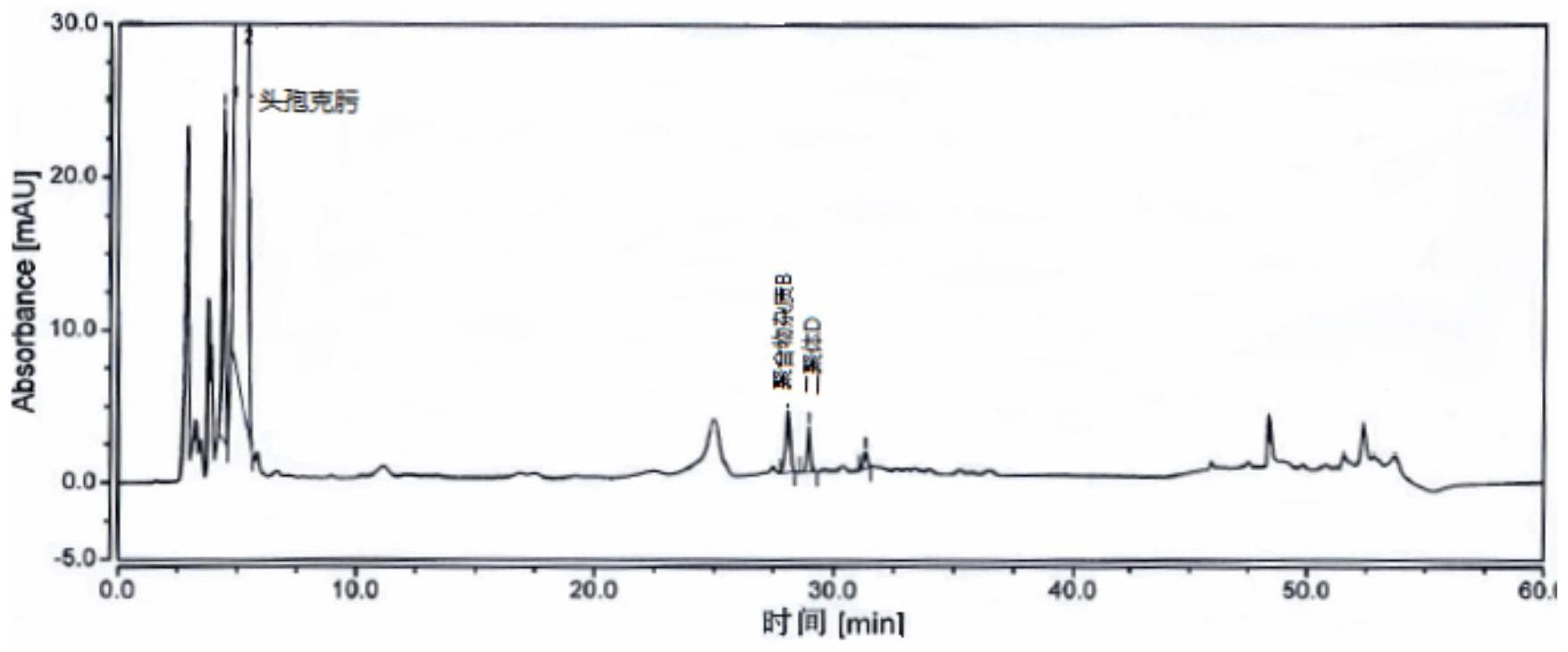
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提供了一种 4 CN 111551654 A 说 明 书 2/10 页 稳定性高、专属性好、可以对多种杂质有效分离进行定性定量分析的检测头孢克肟聚合物 杂质的方法。 本发明的技术方案如下文所示。 本发明一方面提供了检测头孢克肟聚合物杂质的方法,包括使用高效液相色谱法 检测头孢克肟聚合物杂质的步骤,所述高效液相色谱法的检测条件包括: 流动相:由流动相A和流动相B组成,其中,流动相A为甲酸溶液,流动相B为乙腈、甲 醇或乙腈-甲醇;流动相A和流动相B的体积比为20~95:80~5。 任何两种不同的物质,只要它们存在物理、化学或生物学性质上的差异及在不同 物相上的分配系数的差异,便都可以在色谱过程中得到分离、分析或测定。发生在色谱柱中 的分离过程,受热力学因素和动力学因素的控制,而流动相的种类和使用条件既可影响动 力学因素,也可影响热力学因素,对分离过程有非常大的影响。对于头孢克肟聚合物杂质, 现有技术采用分子排阻法测定,并以磷酸盐缓冲体系作为流动相。而本发明人为了便于液 质联用以确证液相色谱条件所测得的杂质是否为聚合物杂质,经过大量的实验开发了甲酸 体系作为流动相,可以很大程度上,分离出聚合物,并增加聚合物杂质和药物成分的分离 度,实现聚合物杂质定性定量的测定。 根据本发明的一些实施方式,所述甲酸溶液的pH范围为2.5~8.0,甲酸的体积浓 度为0.05%~1.0%。优选地,所述甲酸溶液的pH为6.0,甲酸的体积浓度为0.1%。 流动相中合适的pH值,可以抑制溶质的离子化,减少谱带拖尾、改善峰型,提高分 离的选择性等。而离子强度则对于色谱的峰型有一定的影响,如过载、拖尾、峰宽等。本发明 综合考量多种因素,筛选出了流动相适宜的离子强度和pH值。 根据本发明的一些实施方式,高效液相色谱法的检测采用如下梯度洗脱程序: 在0~20min,流动相A的体积百分含量为90~95%,流动相B的体积百分含量为5~ 10%; 在20~28min,流动相A的体积百分含量由90~95%降到80~90%,流动相B的体积 百分含量由5~10%提高到10~20%; 在28~40min,流动相A的体积百分含量为80~90%,流动相B的体积百分含量为10 ~20%; 在40~50min,流动相A的体积百分含量由80~90%降到20~30%,流动相B的体积 百分含量由10~20%提高到70~80%; 在50~51min,流动相A的体积百分含量由20~30%提高到90~95%,流动相B的体 积百分含量由70~80%降到5~10%; 在51~60min,流动相A的体积百分含量90~95%,流动相B的体积百分含量为5~ 10%。 根据本发明的一些优选的实施方式,所述流动相采用如下梯度洗脱程序: 在0~20min,流动相A的体积百分含量为92%,流动相B的体积百分含量为8%; 在20~28min,流动相A的体积百分含量由92%降到80%,流动相B的体积百分含量 由8%提高到20%; 在28~40min,流动相A的体积百分含量为80%,流动相B的体积百分含量为20%; 在40~50min,流动相A的体积百分含量由80%降到25%,流动相B的体积百分含量 5 CN 111551654 A 说 明 书 3/10 页 由20%提高到75%; 在50~51min,流动相A的体积百分含量由25%提高到92%,流动相B的体积百分含 量由75%降到8%; 在51~60min,流动相A的体积百分含量92%,流动相B的体积百分含量为8%。 梯度洗脱是控制流动相的组成,使其在色谱分离过程中发生连续的变化。通过连 续改变流动相中各溶剂组分的比例以连续改变流动相的极性,使每个分析组分都有合适的 容量因子k,从而实现供试品质中各组分在最短时间内的最佳分离。梯度洗脱包括线性梯度 洗脱和非线性梯度洗脱,需要综合考虑流动相的极性、梯度洗脱时间、梯度陡度和梯度洗脱 程序曲线形状等多种因素来确定最终的梯度洗脱程序,其对于待分离组分的保留时间、峰 型等都有非常重要的影响。本发明人梯度洗脱的设计思路以主成分与聚合物杂质以及已知 的聚合物杂质之间分离效果为主,确定了非线性梯度洗脱程序,既能使弱保留溶质之间有 足够的分辨率,也能使强保留溶质以合理的保留时间从色谱柱上洗脱下来,在很大程度上 提高了方法的专属性。 根据本发明的一些实施方式,所述头孢克肟聚合物杂质为头孢克肟聚合物杂质B、 头孢克肟二聚体D、头孢克肟二聚体F和头孢克肟双母核1中的至少一种。 头孢克肟中聚合物杂质的结构未被各国药典收载,也没有相关文献的报道。本发 明采用了一种高比例有机相其适合质谱的方法,充分洗脱样品中可能存在的杂质,并对检 出的杂质采用高分辨质谱进行分子量的确认,同时采用对照品定位,对可能存在的高分子 聚合物杂质进行归属,确定头孢克肟中存在的聚合物杂质为头孢克肟聚合物杂质B、头孢克 肟二聚体D、头孢克肟二聚体F、头孢克肟双母核1中的一种或几种。 根据本发明的一些实施方式,所述高效液相色谱法的检测条件还包括以下i-v中 的一项或多项: i.柱温:30~40℃; ii.色谱柱:十八烷基硅烷键合硅胶柱; iii.流速:0.8~1.2mL/min; iv.检测波长:254nm; v.进样量:5~20μL。 根据本发明的一些优选的实施方式,所述色谱柱规格为4.6mm*250mm,3μm;优选 地,所述色谱柱为Hypersil Gold C18;所述柱温为35℃;所述流速为1.0mL/min。 柱温是一个重要的操作条件,直接影响分离效果和分析速度,对于保留时间、峰面 积、有效塔板数、分离度等都有影响。色谱柱中填料的粒度主要影响填充柱的柱效和背压; 粒度越小,填充柱的柱效越高,在相同条件下,柱效高可提高分离度,但粒度越小,柱压越 大,因此需要配合调整流动相或柱温来降低柱压。在色谱过程中,随着流动相的行进,所携 带的溶质谱带亦一同前行,一方面,溶质在流动相和固定相之间不断地进行着传质过程,同 时,受流动相流型的影响,其浓度分布,即谱带也会发生变化,产生谱带增宽效应。通常,这 种展宽效应与流动相的流速、流型以及溶质在流动相中的扩散速度都有关;流动相的流速 过高会加快洗脱速度,缩短出峰时间,不利于实现分离效果;而流速过低则会降低洗脱速 度,延迟出峰时间,拉宽各组分峰宽,降低检测效率。高效液相色谱检测器的好坏直接关系 到分析结果的可靠性和准确性,不同的仪器参数对于实验结果产生的影响不同,选择合适 6 CN 111551654 A 说 明 书 4/10 页 的检测波长不仅可以提高检测灵敏度,也能有效的改善分离结果。本发明通过综合考量各 种因素,在保证检测的效率、分离度等的情况下,筛选出了最优的参数范围。 根据本发明的一些实施方式,所述检测头孢克肟聚合物杂质的方法,包括以下步 骤中的一步或多步: 1)制备供试品溶液:取供试品加稀释剂超声溶解,并定量稀释为0.2~2.0mg/mL浓 度的溶液;优选定量稀释为1mg/mL浓度的溶液; 2)配制对照溶液:将供试品溶液加入稀释剂溶解并定量稀释为0.2~20μg/mL浓度 的溶液;优选定量稀释为1-10μg/mL浓度的溶液; 3)用高效液相色谱法对供试品溶液和对照溶液进行检测,获得检测结果,根据所 述检测结果定性分析和/或定量计算所述聚合物杂质。 根据本发明的一些实施方式,所述供试品为头孢克肟原料药或包含头孢克肟的组 合物,所述组合物优选为头孢克肟胶囊、头孢克肟颗粒、头孢克肟片、头孢克肟分散片或头 孢克肟混悬剂。 本发明中能够用于头孢克肟原料药及头孢克肟各类剂型药物中的相关聚合物杂 质的检测,检测效果好、适用范围广。 根据本发明的一些实施方式,所述稀释剂选自甲酸、磷酸盐缓冲溶液、甲醇、乙腈 中的至少一种。优选地,所述稀释剂为0.075mol/L无水磷酸氢二钠:0.075mol/L无水磷酸二 氢钠=61:39。 因为头孢克肟的水溶性较差,几乎不溶于水,采用特定的稀释剂可以使头孢克肟 解离,易溶于水,便于测定其中的高分子杂质。 根据本发明的一些实施方式,采用面积归一化法、自身对照法、内标法或外标法进 行高效液相色谱分析;优选采用自身对照法。 本发明中检测结果的分析可以根据实际需求采用合适分析方法。 本发明另一方面还提供一种试剂盒,任选包括稀释剂、甲酸、乙腈。优选地,还包括 对照品。 根据本发明的一些实施方式,所述试剂盒按照如上所述的检测头孢克肟聚合物杂 质的方法来定性或定量检测头孢克肟聚合物杂质。 本发明的有益效果: 本发明提供的检测头孢克肟聚合物杂质的方法能有效检测头孢克肟聚合物杂质, 解决原检测方法专属性差,检测到的杂质绝大部分不是聚合物杂质的问题。 本发明提供的检测头孢克肟聚合物杂质的方法能够在一次检测中同时有效检测 药物中的头孢克肟中聚合物杂质B、头孢克肟二聚体D、头孢克肟二聚体F、头孢克肟双母核1 中的一种或几种,分离度满足定量检测要求,很好的满足原料药和制剂工艺过程中的杂质 监控及原料药和制剂的成品中的杂质控制要求。 附图说明 图1示出了流动相pH值为5.8时的检测谱图; 图2示出了流动相pH值为6.0时的检测谱图; 图3示出了流动相pH值为6.2时的检测谱图; 7 CN 111551654 A 说 明 书 5/10 页 图4示出了采用对比例1中的检测方法得到的检测谱图; 图5示出了采用对比例3中的检测方法得到的检测谱图。











