
技术摘要:
用于在电泳过程中保留分子的盒,其具有外壳,外壳具有配置在其中的泳道。所述泳道具有第一延伸边缘和第二延伸边缘,洗脱模块配置成被接收在泳道中,以将所述泳道划分为第一室和第二室。第一缓冲液池的位置邻近第一延伸边缘,第二缓冲液池的位置邻近第二延伸边缘。洗脱 全部
背景技术:
下一代测序(NGS)正越来越多地用于医学中进行遗传性疾病的诊断以及肿瘤学中 进行合适疗法的选择。由于许多遗传性疾病和可操作癌症基因座是蛋白质编码序列中的单 核苷酸多态性(SNP),大多数临床测序(即Illumina和Ion Torrent平台)是使用短读长测序 组合杂交捕获靶向样品制备进行的(Agilent SureSelect,Roche Nimblegen,IDT xGen)。 然而,短读长NGS方法不适合长程(long-range)基因组分析,例如单倍型测定和结 构变异(“SV”)检测,结构变异在本文定义为涉及长度大于100bp的缺失、重复、倒位、易位、 重复扩增的重排。当SV断裂点和单倍型涉及重复序列区域时,短读长方法尤其不利。 这给临床测序领域带来了重大问题,因为最近对大型患者组的研究表明,一些类 型的癌症包含由SV而不是由SNP所引起的驱动突变(4)。另一项癌症群体研究(全基因组泛 癌分析)发现了52个不同基因座处明显的重复性SV(10)。类似地,人们越来越意识到遗传性 疾病的重要部分(但仍未知)是由短读长方法无法检测到的缺失、重复扩增和其他SV所引起 的(11,12)。最后,有越来越多的证据表明,延伸到整个4mb MHC区域的单倍型结构对于破译 许多复杂的免疫病症来说将是非常重要的(在13中综述)。由此,短读长方法能够识别多态 性的离散点,但它们之间的联系只能通过对家族的分析(间接)推断出来。 SV和扩展单倍型的可靠检测需要长读长的单分子测序(或光学作图方法),例如由 Pacific Biosciences、Oxford Nanopore、10XGenomics、Bionanogenomics和Genomic Vision所开发的方法(2-12)。在短读长基因集合和外显子测序的情况下,出于经济原因,在 临床环境中使用长读长测序很可能仅限于靶向测序。不幸的是,与传统的靶向短读长测序 相比,这些长读长测序方法的靶向样品制备仍然是费时、低效且昂贵的。
技术实现要素:
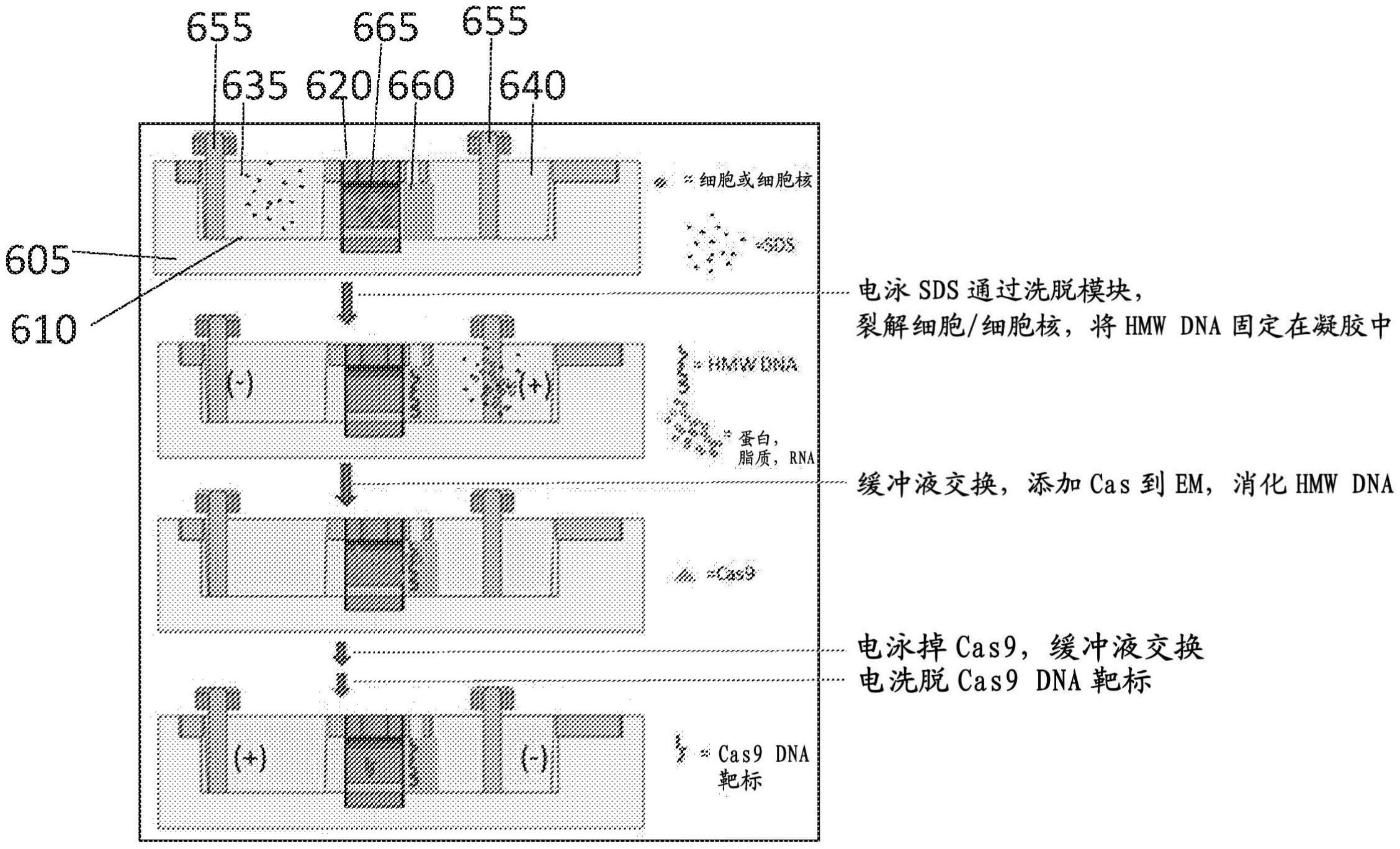
本文描述了各种装置、系统和方法。在一些实施方案中,用于在电泳期间保留分子 的分子保留盒,其包括外壳和配置在外壳内的泳道。泳道可以具有第一延伸边缘和第二延 伸边缘。洗脱模块可配置成被接收在泳道中,并可将泳道划分为第一室和第二室。第一缓冲 液池的位置可邻近第一延伸边缘,第二缓冲液池的位置可邻近第二延伸边缘。洗脱模块朝 向第一室的第一侧可包括多孔无菌滤膜,且洗脱模块朝向第二室的第二侧可包括具有在电 泳期间保留分子的孔径的超滤膜。 在一些实施方案中,该盒还可包括配置在第一室内的至少一个电极和配置在第二 室内的至少一个电极。 超滤膜可以是15kDa超滤器。超滤膜的孔径可以配置成在电泳期间中保留DNA。 5 CN 111742216 A 说 明 书 2/10 页 洗脱模块可位于第一缓冲液池和第二缓冲液池之间的中央处。洗脱模块可位于第 一缓冲液池和第二缓冲液池之间。在一些实施方案中,洗脱模块可包括洗脱模块和样品孔。 洗脱模块可配置为接收样品。 该盒还可包括琼脂糖凝胶。琼脂糖凝胶可紧挨着多孔无菌滤膜浇铸。琼脂糖凝胶 可浇铸成凝胶柱,且凝胶柱的尺寸配置成将进入第一室的靶标分子的损失降到最低。 在一些实施方案中,用于在电泳期间保留分子的分子保留盒包括外壳和配置在外 壳内的多条泳道。所述多条泳道各自具有第一延伸边缘和第二延伸边缘。多个洗脱模块可 各自配置成被接收在多条泳道的一条泳道中,以将每条泳道都划分为第一室和第二室。第 一缓冲液池的位置可邻近每条泳道的第一延伸边缘,第二缓冲液池的位置可邻近每条泳道 的第二延伸边缘。洗脱模块朝向每条泳道第一室的第一侧包括多孔无菌滤膜,且洗脱模块 朝向每条泳道第二室的第二侧包括超滤膜。超滤膜具有在电泳期间保留分子的孔径。 在一些实施方案中,用于分离和收集靶标颗粒的靶标段的方法可包括在洗脱模块 的样品孔中接收样品。可在第一缓冲液室中接收含有SDS的裂解缓冲液,第一缓冲液室沿洗 脱模块的第一侧配置。可施加第一电泳电压将样品组分迁移至沿洗脱模块的第二侧配置的 第二缓冲液室,使靶标颗粒固定在沿洗脱模块的第二侧配置在洗脱模块和第二缓冲液室之 间的凝胶段中,非靶标颗粒通过凝胶段进入第二缓冲液室。可用Cas9反应缓冲液清洗第一 缓冲液室、第二缓冲液室和洗脱模块并填充。洗脱模块可清空并用Cas9酶混合物重新填充, 以切割固定在凝胶段中的靶标颗粒部分。可将SDS终止溶液加载到洗脱模块中,并可施加第 二电泳电压以从靶标颗粒中释放Cas9并将Cas9迁移到第二缓冲液室中。可用洗脱缓冲液清 洗第一缓冲液室、第二缓冲液室和洗脱模块并填充。第三电泳电压可以相反方向施加,将靶 标颗粒的切割部分从凝胶段迁移到洗脱模块中。 洗脱模块的第一侧可包括超滤器,洗脱模块的第二侧可包括多孔无菌过滤器。在 施加第三电泳电压期间和之后,多孔无菌过滤器可防止靶标颗粒的切割部分离开洗脱模 块。 靶标颗粒可以是DNA,且DNA的切割部分可包含期望的基因组靶标。 在一些实施方案中,施加第二电泳电压可短于第一电泳电压和/或第三电泳电压。 施加第一电泳电压可使SDS迁移通过洗脱模块以裂解样品,并包被样品的非靶标 颗粒,从而使非靶标颗粒通过凝胶段而进入第二缓冲液室。施加第二电泳电压将小于靶标 颗粒的颗粒迁移到第二缓冲液室中。 电泳仪器系统可包括电泳工作站和抽屉,将抽屉配置为接收至少一个电泳盒,诸 如本文所述的电泳盒。系统还可包括液体处理机器人和横向延伸臂,将横向延伸臂配置成 横向移动抽屉,使得在第一横向方向上移动抽屉而将至少一个盒的第一侧暴露于液体处理 机器人,以及在第二横向方向上移动抽屉而将抽屉插入电泳工作站。电泳部分容纳对应于 至少一个盒的电极,其中将电极配置成施加电泳电压。系统还可包括至少一个冷藏小室和 至少一个室温储藏小室。 附图说明 图1A显示了根据一些实施方案的SageHLS盒。 图1B显示了根据一些实施方案的SageHLS仪器。 6 CN 111742216 A 说 明 书 3/10 页 图2显示了根据一些实施方案用于分离大基因组DNA靶标的HLS-CATCH工作流程。 图3显示了根据一些实施方案由HLS-CATCH制备的200kb BRCA1片段的靶向10X Genomics测序。 图4显示了根据一些实施方案来自图3的BRCA1序列经过10X Long Ranger软件处 理后的全相位输出。 图5显示了根据一些实施方案使用10X Genomics/Illumina测序对HLS-CATCH靶标 进行SV检测。 图6显示了根据一些实施方案的紧凑型多通道CATCH-1D盒样机的初步概念。 图7显示了根据一些实施方案的CATCH-1D工作流程。 图8显示了根据一些实施方案的示例性流程图。 图9显示了自动CATCH-1D仪器可能的液体处理器平台布局示例,说明了根据一些 实施方案将电泳工作站集成到液体处理机器人的平台上,其中尺寸指典型的小型OEM液体 处理工作台的占地空间(footprint)。 具体实施方案 开发了用于靶向性长读长测序的集成化高通量自动化样品制备系统。所提出的系 统旨在于临床诊断环境中进行强大的全自动处理。 提供了半自动研究仪器系统(图1A-B),用于提取和酶处理极高分子量(HMW)DNA (100-2000kb)。该系统使用完整的细胞或分离的细胞核作为输入样品。将输入样品加载到 琼脂糖凝胶盒(图1A)中,通过SDS电泳经由样品孔小室从样品中提取染色体长度的DNA。SDS 包被的蛋白质、脂类通过中央琼脂糖凝胶柱电泳离开样品孔,但染色体长度的DNA牢固地缠 绕并固定在样品孔的琼脂糖凝胶壁上。样品孔可以清空和重新填充,而不会丢失任何DNA。 这允许通过用酶反应混合物重新填充样品孔来处理固定化的DNA。许多常用的DNA处理酶容 易扩散到琼脂糖中,包括许多限制性酶、DNA聚合酶、连接酶、转座酶、非特异性DNA切割酶和 化脓链球菌(S.pyogenes)Cas9。DNA处理后,进行再一轮大小选择电泳,然后将DNA产物电洗 脱到沿着凝胶分离柱的一侧排列的一系列缓冲液填充的六个洗脱模块中(图1A)。DNA处理 步骤包括一些切割,以将所需DNA产物的长度减小到2兆碱基(mb)以下——大于2mb的DNA将 保持固定在样品孔中,在电泳过程中无法移动。 如图1A所示,每个盒有两个物理分开的样品处理区。该盒具有标准的96孔板空间。 中央琼脂糖通道具有两个加样孔。将细胞或细胞核加载到样品孔中,并将基于SDS的裂解试 剂加载到试剂孔中。进行电泳以驱动SDS穿过样品孔小室,在那里将细胞或细胞核裂解。染 色体大小的基因组DNA固定在样品孔壁上,而其他组分则与SDS一起被带到底部电极室。DNA 处理和大小选择电泳后,DNA产物被电洗脱到位于琼脂糖通道右侧的六个洗脱模块阵列中。 图1B显示仪器约1英尺宽/高/深,可容纳两个盒。总样品处理量为4个样品,运行时 间在3到7个小时之间变化,这取决于大小范围和大小选择步骤所需的分辨率。 SageHLS系统能够与定制的Cas9核酸酶一起使用,以分离特定的大基因组DNA靶 标,其能够有效地用于在10X Genomic Chromium系统中制备靶向性DNA文库。图2显示了工 作流程的示意图,本文称之为“CATCH”。将完整的细胞或细胞核加载到盒中,并按上述进行 电泳提取,使染色体长度的基因组DNA固定在样品孔壁中。酶法DNA处理是使用定制的Cas9 7 CN 111742216 A 说 明 书 4/10 页 切割酶来进行的,这些切割酶被设计成切割待测序的一个或多个基因组区域之外的区域。 Cas9消化后,进行大小选择电泳,使Cas9切割的靶标区远离样品孔,并远离保持被截留在样 品孔壁中的未切割基因组DNA。经凝胶通道电洗脱后,通过qPCR对产物进行定位,并直接用 于10X Chromium系统中进行文库构建。 对包含人类BRCA1基因的200kb基因组区域所进行的靶向测序总体工作流程已经 得到证明。以150万人二倍体培养细胞作为输入材料,从峰洗脱模块中回收了约30,000-50, 000拷贝的BRCA1片段,如通过Taqman qPCR所测得的相对非靶向性对照基因(RNaseP)约25 倍的富集度。在10X Genomics Chromium系统上制备文库后,在Illumina NextSeq500系统 上对它们进行测序。该运行产生了155X的靶向性BRCA1区覆盖率,只有4.4倍的非靶向性基 因组剩余部分的覆盖率(图3)。使用10X Long Ranger比对软件进行的进一步分析证明,全 相位单倍型能够确定两个等位基因(图4)。根据gRNA设计预测,超过一半的BRCA1 HLS- CATCH产物都是全长靶标。 在平行实验中,已经证明HLS-CATCH靶向也能用于检测大SV,因为在研究充分的细 胞系GM12878中设计了靶向40个已知SV的gRNA。靶标区是在单个多重HLS-CATCH方法中分离 到的,其靶向40个不同的100kb基因组片段。通过qPCR将靶标定位在HLS盒输出中,并对 BRCA1基因进行10X Genomics文库制备和Illumina测序。证实在GM12878 chr1上检测到纯 合的40kb缺失的一些初步数据示于图5中(Shin,Ji,手稿准备中)。 图3-5的数据表明,HLS-CATCH与10X Genomics文库构建方法组合能够以靶向性方 式提供高质量的长程单倍型和SV识别(calls)。我们相信这是第一次成功证实了使用大于 20kb的基因组片段所进行的靶向性测序工作流程(14)。组合技术的成功来自于HLS-CATCH 方法的高效靶标富集与10X Chromium工作流程的整合性扩增 文库构建方法之间的出色互 补性。 基于上述HLS-CATCH/10X Genomics工作流程的诊断测试可以在基因测试和肿瘤 学方面具有令人信服的优势。该工作流程的靶向性大大降低了Illumina测序成本(用于 100X相位Chromium全基因组测序的ILMN试剂为$8000,而用于图4中BRCA1的100X相位靶向 性Chromium覆盖率的ILMN试剂为$500)。此外,对大的靶向片段的分析提供了在同一测定中 同时检测SV和SNP的机会,而且还增加了单倍型确定的好处。 方法 用于这些应用的系统具有高样品处理量可以是有利的,以便增加和/或最大化所 处理的样品数量。此外,以可重复的方式实现高效、特异的Cas9靶标切割可以是有利的。在 用小鼠白细胞(WBC)和人培养细胞(淋巴母细胞和HEK293细胞)进行的大约30次HLS-CATCH 实验中,CATCH靶标回收率差异很大,在2%到50%的范围内。类似地,靶标富集度在15到200 倍变化。幸运的是,用这些范围下限的样品进行的10X Chromium文库制备也运行相当好。例 如,图3-5的高覆盖率靶标数据是用仅约为1.5%(50,000个拷贝)的总体CATCH靶回收率、约 为25倍的富集度(这两个值均为由qPCR相对于RNaseP基因拷贝数所测量的)所产生的。这可 以是HLS-CATCH/10X Genomics工作流程的另一补充特征:整体工作流程的成功取决于大量 富集,而不取决于绝对靶标纯度,在后一种情况下,可能由于“条码冲突”而导致无效,在“条 码冲突”的情形下,多个等位基因在液滴中共同定位并被标记上相同的linked-read条码。 改进和扩充HLS-CATCH以处理极低的细胞/细胞核输入也可以是有利的。这是进行 8 CN 111742216 A 说 明 书 5/10 页 基因分型诊断样品(如活检样品)的关键问题,这些样品可以是非常有限的大小和细胞数 量。如上文所讨论的,成功使用低细胞/细胞核输入将取决于能否成功优化Cas9消化条件。 此外,充分评价和扩充HLS-CATCH使用各种组织类型的能力可以是重要的,所述组 织类型包括血沉棕黄层、冷冻血液、新鲜和新鲜冷冻的实体组织,包括各种类型的肿瘤活检 材料。 在阶段I,可以设计盒来完成HLS-CATCH大靶标富集。新的设计可以取消原始HLS盒 的二维电洗脱步骤,由此允许制造每个盒能够处理6到12个样品的盒(在96孔板空间中)。新 的盒类型在本文被称为“CATCH-1D”,其中1D代表“一维”。 阶段I可以包括使用750,000个二倍体哺乳动物细胞(人类或小鼠)的样品,以在 CATCH-1D样机中实现单拷贝的200,000bp基因组靶标片段至少20%(300,000个拷贝)的回 收率。可通过qPCR测量拷贝数量。200,000bp基因组靶标的富集至少是非靶向基因组序列背 景的20倍,可通过qPCR测量富集。此外,可以证明CATCH-1D样机产生的CATCH靶标能够在10X Genomics Chromium/Illumina工作流程中被高效地测序。 阶段II可以包括具有液体处理能力的CATCH-1D仪器的开发,以进行高通量全自动 CATCH靶标制备。可以针对CATCH-1D操作优化Cas9消化。阶段II还可以包括调整CATCH-1D盒 或工作流程以用于低的细胞或细胞核输入,并调整CATCH-1D盒或工作流程以用于诊断上重 要的组织类型。 阶段I方法 CATCH-1D盒及其操作的基本概念如图6和图7所示。SageHLS盒(图1A)经过改进,以 便外壳605的每条泳道610都包括一对位于洗脱模块620任一侧的缓冲液池635、640。每条泳 道610都在配置为接收洗脱模块615的槽的任一侧具有延伸边缘615。当洗脱模块620插入相 应的槽中时,泳道被划分为第一室625和第二室630。第一室625可以包括第一缓冲液池635, 第二室630可以包括第二缓冲液池640。 在一些实施方案中,洗脱模块620位于中央位置,大致位于中央位置或者位于缓冲 液池635、640之间。在洗脱模块620的一侧,连接有多孔无菌过滤膜645。将一小段琼脂糖凝 胶660浇铸在无菌过滤器645的外表面上。在洗脱模块620的另一侧,连接有超滤膜650。超滤 器650具有在电泳期间会保留DNA的孔径。 在CATCH-1D工作流程中,洗脱模块620同时充当样品孔665和洗脱模块620。如图7 所图示说明的和图8的流程图所示的,将细胞/细胞核样品加载到洗脱模块620中(810),清 空左缓冲液室635并用含有SDS的裂解缓冲液重新填充(815)。施加电泳电压(820),例如通 过一个或多个电极655施加,以便SDS能够迁移通过洗脱模块、裂解输入材料并包被蛋白质 和其他非DNA细胞组分,以便它们能够迁移通过小凝胶段660并进入右缓冲液室640中。在此 期间,基因组DNA迁移到小凝胶段660中并被固定在那里,如原始HLS方法一样(原始方法如 图2所示)。此时,中止电泳,彻底清洗盒的所有三个小室(左右缓冲液室635、640和洗脱模块 620)(825),并用Cas9反应缓冲液重新填充(830)。然后清空洗脱模块620(835)并用Cas9酶 混合物重新填充(840),Cas9酶混合物将特异性切割来自固定在凝胶660中的染色体长度基 因组DNA的所需基因组靶标。消化后,向洗脱模块620加载SDS终止溶液(845),并进行电泳 (850),以便Cas9能够从DNA中释放并移动到右缓冲液室640中。这种清除所需的电泳时间非 常短,因为在琼脂糖凝胶660中变性的Cas9蛋白的迁移要比大DNA CATCH靶标快得多。该 9 CN 111742216 A 说 明 书 6/10 页 Cas9清除电泳后,清洗盒(855)并用洗脱缓冲液重新填充(860)。以相反方向进行电泳(865) 以将CATCH产物从小凝胶段660移动到洗脱模块620中,其中位于洗脱模块620左侧的超滤膜 650防止靶标逃离。 CATCH-1D盒的设计比原始SageHLS盒简单得多,能够进行样机制作。能够使用具有 定制电极阵列的Sage Science PippinHT仪器(如附录A所示)来进行CATCH-1D样机测试。 PippinHT仪器能够被改进以接收具有多至12个通道的盒。所有液体处理步骤都可以手动进 行(例如在阶段I),也可以自动进行(例如在阶段II)。 与原始HLS工作流程不同的CATCH-1D工作流程中存在多种试剂污染风险。一个例 子来自使用洗脱模块作为输入样品孔。输入样品的一些组分可能粘附在洗脱模块表面,表 明在裂解和清洗步骤中很难去除。类似地,在初始裂解或Cas9清除步骤之后存在的风险是 在洗脱模块中可能会留下一些残余量的SDS。(原始HLS盒的洗脱模块不暴露于输入样品或 浓缩SDS中)。 另一可能的挑战是调整凝胶柱的尺寸,以便用SDS就能够完成电泳Cas9清除,而不 会丢失进入右缓冲液池的CATCH靶标。预计对于大于100kb的CATCH靶标来说,这不会是严重 的挑战,并能够通过qPCR测量右室缓冲液的靶标损失(在Cas9清除电泳之后)。 CATCH-1D工作流程的另一重大风险是缺少尺寸选择电泳步骤。在原始HLS-CATCH 工作流程中,正好在洗脱前进行大小选择电泳以增加CATCH产物的纯度。所提出的CATCH-1D 工作流程则省略了尺寸选择电泳,尽管电泳Cas9清除步骤提供了除去迁移明显快于CATCH 靶标DNA的低分子量DNA的可能性。在任何情况下,一些非特异性切割的HMW DNA很可能会在 最后的洗脱中与CATCH产物一起被洗脱,CATCH产物的纯度就不会像HLS-CATCH工作流程中 那样高。然而,这可能没有问题,因为用适度的富集倍数(约15-25倍)就能够获得优良的10X 测序结果。此外,进一步优化Cas9的消化条件、更好的gRNA设计以及新的突变Cas9酶(或类 似的可编程内切酶)都可能降低大小选择的重要性。 阶段II方法 在阶段I,CATCH-1D系统的可行性已得到证明。阶段II包括全自动高处理量CATCH- 1D系统的开发。 CATCH-1D盒和仪器开发 阶段II的特点是在自动化CATCH-1D系统中集成液体处理功能和电泳的详细系统 工程研究。图9说明了示例性的自动化仪器。电泳工作站直接集成在小型OEM龙门式 (gantry-style)液体处理机器人的平台上。电泳工作站可以具有可容纳四个CATCH-1D多通 道盒的电动抽屉。抽屉能够横向延伸,以露出盒顶部用于液体处理步骤(加载样品/试剂,清 洗盒,取出产物),并退回到电泳工作站(其容纳有电极)内部用于电泳步骤。单个液体处理 头就可以是足够的,LH通道的数量与每个盒的通道数量相匹配(在图9中,我们展示了每个 盒8个通道)。如图9所示,可为输入样品和Cas9试剂提供冷藏(4℃)空间,为裂解试剂和电泳 缓冲液提供室温存储空间,为一次性吸头提供存储空间,并为使用过的吸头和废液提供废 物室空间。 可以制造样机型多通道CATCH-1D盒,其可包括确定每个盒所处理的最佳样品数量 和样品输入大小。此外,可以确定作为通道/洗脱模块尺寸的函数(如下所解释的)的DNA提 取和Cas9消化的效率。CATCH-1D盒的通道可以很小,以便能够在每个盒中处理许多样品。也 10 CN 111742216 A 说 明 书 7/10 页 可以决定确保提取出足够多的CATACH靶标,以获得对于诊断使用而言足够的10X Genomics linked-read覆盖率。 在评价通道尺寸对提取和CATCH回收的影响之后,还可以构建第二个实质上优化 的CATCH-1D盒。也可以构建能够用第2版盒进行全自动化测试的初始仪器样机。也可以开发 生产版本的CATCH-1D盒和自动化仪器。可确定盒和仪器的最终生产版本的制造规程,并可 对最终系统进行测试。 CATCH-1D中HMW DNA提取的优化 CATCH-1D方法的总体效率是两个值的综合。一是从输入样品中提取初始基因组 DNA的效率。二是Cas9消化的CATCH靶标的回收效率。据信CATCH-1D中初始基因组DNA提取的 效率是下述的函数:1)发生细胞裂解和DNA固定的琼脂糖凝胶样品孔的表面积(表面积(SA) 越大越好),以及2)提取过程中每个细胞所使用的洗涤剂的量(越多越好)。为了检验这一问 题,可以用不同的琼脂糖凝胶横截面积来制作多通道CATCH-1D样机,并可将提取效率作为 样品输入的函数进行评价。提取效率可通过频繁切割限制酶(frequent cutting restriction enzyme)来消化固定化DNA并电洗脱消化物后的总DNA回收来测量。长基因组 DNA片段(100-200kb)的提取效率可通过用稀有切割限制酶(rare-cutting restriction enzymes)来消化固定化的DNA并测试已知长度的特定基因组DNA限制性片段的电洗脱产量 来测量。特异性基因组DNA片段的回收率可通过qPCR测量。评价的最大输入量可以是750, 000个二倍体人类细胞,因为阶段I工作可证明该输入将产生对于10X Chromium测序工作流 程中的高覆盖率而言足够的CATCH靶标(至少300,000个拷贝)。 Cas9消化及CATCH靶标回收的优化 一旦理解了提取效率与通道/洗脱模块横截面的比例关系,就可以开始研究Cas9 消化效率,以确定其如何随输入样品量和通道尺寸成比例变化。由于许多类型的临床样品 都会有少量的输入细胞,因此可以在低细胞输入下评价和优化盒样机的性能。在单个靶向 CATCH实验中,仅回收少至50,000个靶标就有可能得到良好的10X Genomics序列覆盖率(约 100X)。在几个实验中,我们已经实现了Cas9靶标(200kb的BRCA1基因座靶标)的50%回收 率。在这个回收值下,仅50,000个完整细胞(或细胞核)的输入就足以达到等价的(~100X) Chromium文库测序覆盖率。当达到更高的靶标回收率时,所需的细胞输入将按比例降低,这 可能很重要,因为许多临床相关样品会具有极低的细胞输入。使用低至10,000个的人类细 胞输入,可实现100X的靶向测序覆盖率。 除了优化适当的盒通道尺寸进行有效提取和CATCH靶标回收外,还可以讨论gRNA 设计、gRNA类型、Cas9酶的选择以及盒内Cas9反应条件。 Cas9反应条件(酶浓度和缓冲液)是进行CATCH靶标回收的主导因素——gRNA类 型、gRNA设计和Cas9类型具有较小的影响。几乎每个所测试的gRNA都在一定程度上对其预 期靶标起作用,75%的引导显示PCR产物在等摩尔靶标/Cas9比率下(浓度在0.1-0.4nM范围 内)完全消化。此外,许多不同的gRNA设计工具(guidescan.com,位于MIT的张峰实验室网 站,安捷伦的SureDesign工具)都可成功使用。此外,使用两部分合成的gRNA和T7pol体外转 录的单个gRNA可以看到类似的切割效率(尽管我们觉得使用合成的RNA更容易和更可靠)。 在我们的体外测定中,还没有发现可商购的Cas9突变体比野生型Cas9酶更具特异性。 尽管有这些初步结论,但CRISPR核酸酶家族仍在不断扩大,可能改造出可对我们 11 CN 111742216 A 说 明 书 8/10 页 的方法有用的新突变体。因此,可以在CATCH-1D盒中筛选增强性能的更多酶。类似地,也可 以测试最近可商购的全长合成单引导RNA。单引导可简化Cas9装配工作流程。例如,可以使 用Synthego,因为供应商已经宣传他们的合成单引导与体外转录的单gRNA相比具有显著提 高的特异性。 在HLS-CATCH方法中,每个切割部位窗口使用2-3个gRNA(每个切割部位使用多个 引导,以避免单个gRNA识别序列中的SNP会消除切割部位的可能性),切割部位窗口约为 2kb,样品孔中总浓度为1-4μM的Cas9酶(以Cas9:gRNA总浓度的1:1比例组装),已经实现最 佳的靶标覆盖率。在SageHLS方法中,通过电泳“注射”Cas9酶到固定HMW DNA的样品孔壁中, 靶标回收率也显著提高。在约50V下使用1分钟时间的电泳。酶浓度和电泳注射是对CATCH靶 标产量具有最大影响的两个参数。 用于其他临床样品类型的CATCH-1D方法的开发 HLS工作流程可适用于各种材料,包括从上述所有来源获得的全血、冷冻全血、血 沉棕黄层、新鲜实体组织、新鲜冷冻组织、肿瘤活检物和细胞核。对于CATCH方法而言,所提 取的DNA几乎是全染色体长度或者大小>>2mb,对于在琼脂糖凝胶中发生高效固定可能是至 关重要的。这意味着CATCH方法可以对新鲜材料或由新鲜材料制备的细胞核最有效。这也意 味着CATCH方法不太可能对福尔马林固定的组织起作用。 SageHLS开发是用哺乳动物白细胞(WBC)或人类细胞系或从这些来源分离出的细 胞核来完成的。SageHLS系统已用于从~50μl的全血中分离出HMW DNA,其中应含有约2.5μg 的基因组DNA,这对于CATCH方法是足够的输入。此外,已经成功地使用SageHLS对冷冻的人 组织培养细胞(HEK293)进行HMW DNA分离。根据这两块数据,可以找到处理血沉棕黄层、冷 冻的血沉棕黄层和冷冻的全血的操作规程。 对于组织操作而言,细胞用细胞仪和FACS仪器(BD Biosciences和Miltenyi Biotech)的供应商已开发了几种用于组织解离的仪器 试剂系统。这包括机械和酶处理的 组合,然后进行选择性过滤,以产生单个细胞或细胞核的悬浮液。这些系统可在使用新鲜材 料进行CATCH-1D输入时运行良好。 基于冷冻组织培养细胞的有限成功率,CATCH最可行的临床组织样品将是新鲜冷 冻组织。可以集中努力在这个样品类型上,并可开始使用上述BD和Miltenyi仪器系统进行 研究。 CATCH-1D试剂盒开发的一致性可重复工作流程 CATCH-1D 10X Genomics工作流程可在临床测序的许多领域有着广泛的应用。为 此,CATCH-1D系统的开发和优化包括建立快速生产新CATCH试剂盒的最佳开发路径。这可以 包括整合用于基因组切割位点选择的生物信息学、切割位点窗口内的gRNA选择、用于gRNA 订购和QC的流线型管道、以及可跟踪的产品编号、库存和包装。 可以对临床测序市场进行调查,以选择可以受益于CATCH-1D方法的关键诊断领 域。例如,CATCH-1D可用于在MHC区实现长程测序。此外,在遗传性疾病检测和肿瘤学方面还 有其他吸引人的测定领域。 可以提供优化的gRNA设计工作流程,其能够用于创建人类基因组的任何区域(或 一组区域)的CATCH测定。遗传性疾病检测中的一些测定可能只涉及单个基因以及侧翼区 域。其他检测可能涉及通常与SV有关的一组癌症基因(可能低至100个)(10)。还有一些可能 12 CN 111742216 A 说 明 书 9/10 页 涉及测定设计,其产生跨越广泛基因组区(如MHC)的CATCH产物(14)。 可以评估专为分析靶向性CATCH-1D 10X Chromium测序结果而设计的10X Genomic Long Ranger软件的修改、定制或新版本。 可以开发用于验证新试剂盒性能的10X Chromium测序工作流程和QC方法。尽管 qPCR是廉价的快速评估回收率和富集度的方法,但可以开发临床测序方法。 本申请中所提及的任何和所有出版物或其他文献,包括但不限于专利、专利申请、 文章、网页、书籍等,均通过引用以其整体并入本文。 本文已描述了装置、系统和方法的示例性实施方案。如其他地方所述,这些实施方 案仅为说明性目的进行描述而不是限制性的。其他实施方案是可能的,并被本公开所涵盖, 这将从本文所包含的教导中显而易见。因此,本公开的广度和范围不应受到上述任何实施 方案的限制,而应仅根据由本公开及其等效者所支持的权利要求书来定义。此外,本公开的 实施方案可包括方法、系统和装置,其还可包括来自任何其他公开方法、系统和装置的任何 和所有元件,包括与分子处理相应的任何和所有元件。换言之,来自一个或另一个公开实施 方案的元件可以与来自其他公开实施方案的元件互换。此外,所公开实施方案的一个或多 个特征/元件可以被除去且仍然得到可专利性主题(并由此而产生本公开的更多实施方 案)。相应地,本发明的一些实施方案可以通过具体缺少在此类现有技术中公开的系统、装 置和/或方法的一个或多个元件/特征而与一个和/或另一个参考/现有技术在可专利性上 明显不同。换言之,某些实施方案的权利要求可以包含否定限制,以明确排除一个或多个元 件/特征,而产生在可专利性上明显不同于包括这些特征/元件的现有技术的实施方案。 附录A 13 CN 111742216 A 说 明 书 10/10 页 14 CN 111742216 A 说 明 书 附 图 1/9 页 图1A 图1B 15 CN 111742216 A 说 明 书 附 图 2/9 页 图2 16 CN 111742216 A 说 明 书 附 图 3/9 页 图3 17 CN 111742216 A 说 明 书 附 图 4/9 页 图4 18 CN 111742216 A 说 明 书 附 图 5/9 页 图5 19 CN 111742216 A 说 明 书 附 图 6/9 页 图6 20 CN 111742216 A 说 明 书 附 图 7/9 页 图7 21 CN 111742216 A 说 明 书 附 图 8/9 页 图8 22 CN 111742216 A 说 明 书 附 图 9/9 页 图9 23











