
技术摘要:
本发明公开了一种硅炔杂化树脂、固化物、陶瓷材料、复合材料及制备方法。其为式I聚合物或者无规共聚物:式I如下所示,n=1~20,R1或R2独立地为氢或甲基;Ar为或无规共聚物由通式A、B和C中的两种或三种表示的结构单元组成,数均分子量为500~1800,封端基为‑Ar‑C≡CH 全部
背景技术:
20世纪50年代以来,航空航天、电子工业和交通运输等领域的发展对材料的耐热 性能的要求越来越高,许多耐热高分子材料相继研发并走向应用。有机无机杂化材料在新 型功能材料中有广阔的应用前景,是20世纪90年代末化学和材料学科中的热门研究领域。 芳基乙炔树脂是一类新型的耐高温热固性树脂,这类树脂为液态或易溶易熔的固 态,固化时无小分子逸出,固化后呈高度交联结构,且由大量刚性芳环,因而耐热性和残炭 率高,有望在热防护材料中发挥作用。在芳基乙炔中引入硅原子的硅芳炔有机-无机杂化树 脂的加工性能和力学性能优于芳基乙炔树脂,高温陶瓷化中会有高热稳定性和高拉伸强度 的碳化硅陶瓷形成。 Barton研究小组报道了(Journal of Polymer Science:Part A:Polymer Chemistry,1990,28:955-965.)由三氯乙烯制备乙炔与硅烷的聚合物,用正丁基锂将三氯 乙烯定量转为乙炔基二锂与二氯硅烷缩聚得到了高分子量的聚合物,可拉成纤维和浇注为 薄膜,热解后形成碳化硅。其主要目的在于研究如何提高硅炔树脂的分子量,树脂固化温度 较高,热解过程中耐热结构少,烧结中树脂链的断裂而导致硅质量分数在烧结物中的降低。 虽硅炔聚合物分子量的增加有利碳化硅的制备,但是分子量提升前后的树脂的热解陶瓷化 率并未提高。 Corriu研究团队则报道了(Organometallics,1992,11:2507-2513.)将二乙炔基 (丁二炔基)结构引入硅炔聚合物主链结构中,丁二炔基是活性基团,加热可使硅炔树脂交 联,形成网络结构后再热解,虽然其陶瓷化率高达84%,但是其树脂固化温度较高(400℃)、 加工窗口较窄。 周权等人(Polym Int,2014,63:1531-1536.)由乙炔基溴化镁与二氯硅烷反应制 得二乙炔基硅烷,再格氏化后与二氯硅烷合成了硅炔树脂,但树脂由差示扫描量热法(DSC) 测得的起始固化温度高于200℃。而且树脂结构中含有Si-H结构,室温下易发生硅氢加成而 引起固化交联反应失去树脂的可加工性,所以需低温保存树脂(树脂室温贮存稳定性差), 一旦开始用于加工复合材料,可加工时间短,需快速用完,否则易交联失去可加工性。 中国专利文献CN103333341A公开了一种耐高温杂化硅氮烷树脂及其制备方法,由 二乙炔基苯与二氯硅烷缩聚而成,其封端用的是氨基苯乙炔,电子通过硅-氮键形成整个分 子链的离域性小,会影响树脂的固化和性能,也会影响树脂中硅含量,降低陶瓷中碳化硅陶 瓷的质量分数。而且该树脂的固化和性能未有数据公开。 中国专利文献CN108752374A公开了一种乙炔基苯基封端的含硅芳炔基炔丙基醚 树脂及其合成、三元树脂及其制备、复合材料及其制备,其采用双酚A二炔丙基醚制备得到 树脂材料,该树脂材料由于含氧导致耐热性能低(二氧化硅陶瓷耐热低于碳化硅陶瓷),硅 含量(树脂中硅质量分数)低于10%,陶瓷化后碳化硅陶瓷含量低,陶瓷性能(热性能和力学 6 CN 111548497 A 说 明 书 2/16 页 性能)较低。 中国专利文献CN104962085A公开了一种提高含硅芳炔树脂基复合材料力学性能 的方法,说明书41段提及了PSA的分子结构式,其采用共聚合单体是间二乙炔基苯,树脂中 硅含量(质量分数)只有14%左右,陶瓷化后碳化硅陶瓷质量分数低,陶瓷性能较差。 另外,现有的硅芳炔树脂中硅的质量分数低(约14%左右),陶瓷化后碳化硅陶瓷 含量低,陶瓷的热性能和力学性能低。
技术实现要素:
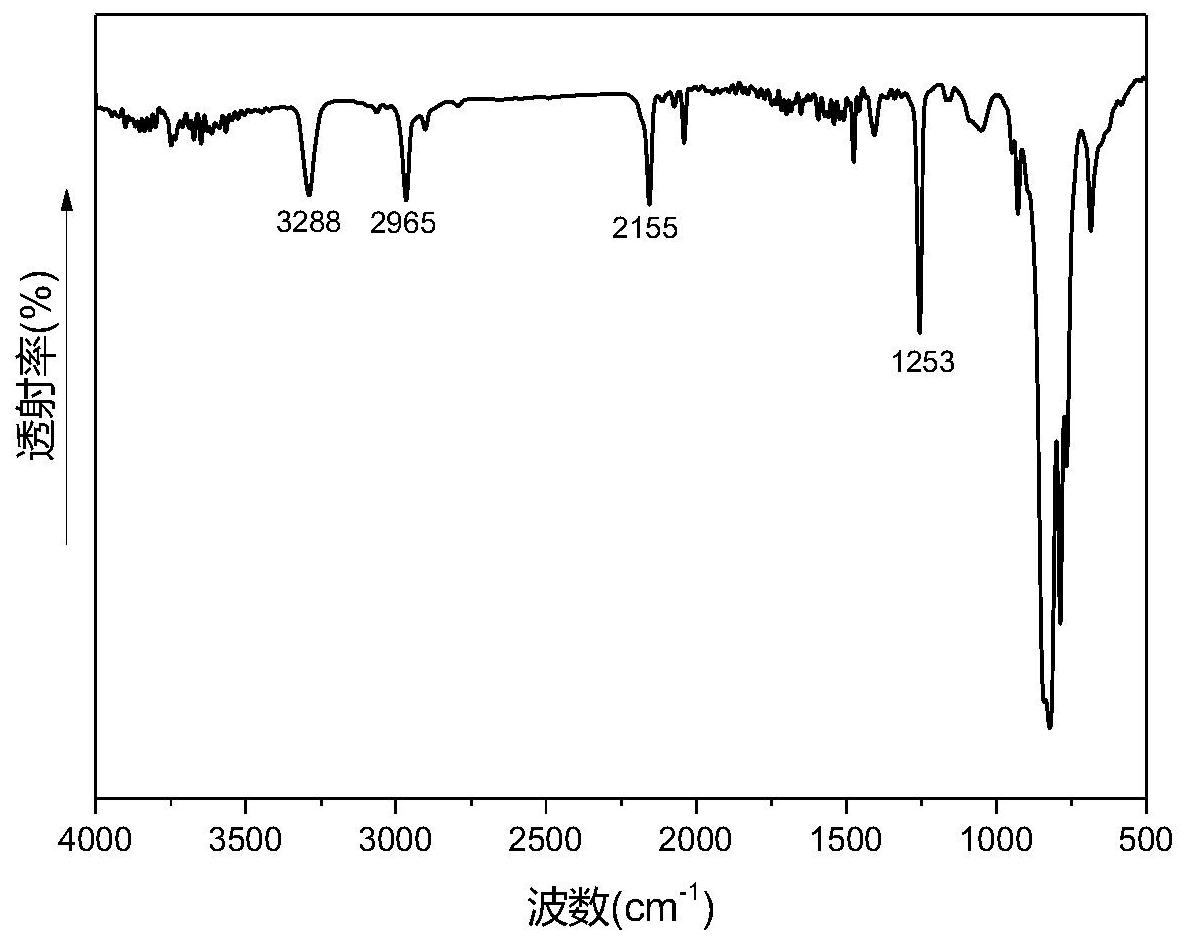
本发明所要解决的技术问题是克服了现有技术中制备碳化硅陶瓷前驱体聚合物 (树脂)室温贮存稳定性差、加工工艺性差、固化交联温度高,硅含量低,以及陶瓷化碳化硅 陶瓷含量低等的缺陷,提供了一种硅炔杂化树脂、固化物、陶瓷材料、复合材料及制备方法。 本发明的硅炔杂化树脂可加工区间较宽,易加工,可避免现有碳化硅陶瓷烧制工艺对成型 的限制;硅炔杂化树脂中硅的质量分数较高,使得陶瓷化后陶瓷中碳化硅质量分数的提高, 进而提升了陶瓷的热性能和力学性能。 本发明通过巧妙的结构设计,采用现有的化学方法合成出甲硅烷与乙炔交替共聚 形成的可陶瓷化硅炔杂化树脂,该树脂中,硅原子上的σ电子与乙炔的π电子会发生电子离 域而形成σ-π共轭聚合物,即整个分子链形成σ-π共轭结构,π-电子密度沿着主链离域,电子 实现在整个分子链的离域带来不同的性能,该聚合物的光学效应会红移,可作为潜在的半 导体、光电荧光材料和陶瓷前驱体。 为实现上述目的,本发明采取的技术方案是: 本发明提供了一种可陶瓷化硅炔杂化树脂,所述可陶瓷化硅炔杂化树脂为化学结 构为式I的聚合物或者无规共聚物: (1)式I的聚合物: 其中,n=1~20,R1、R2独立地为氢或甲基;Ar为 (2)无规共聚物: 所述无规共聚物由通式A、通式B和通式C中的两种或三种表示的结构单元组成,所 述无规共聚物的数均分子量为500~1800:所述无规共聚物的封端基为-Ar-C≡CH,所述Ar 为 7 CN 111548497 A 说 明 书 3/16 页 所述式I的聚合物中,所述n较佳地为2~18,更佳地为4~16,例如5、8、10、12、15或 者16。 所述式I的聚合物中,较佳地,所述R1或者所述R2为甲基。 所述式I的聚合物中,较佳地,所述Ar为 较佳地,所述式I的聚合物的数均分子量Mn为500~1800,更佳地为800~1700,例 如1360、1570或者1650。 所述无规共聚物中,较佳地,所述无规共聚物由通式A和通式B表示的结构单元组 成,或者通式A和通式C表示的结构单元组成,或者通式B和通式C表示的结构单元组成,或者 通式A、通式B和通式C表示的结构单元组成,更佳地为由通式A和通式B表示的结构单元组 成。 当所述无规共聚物由通式A和通式B表示的结构单元组成时,通式A和通式B的摩尔 比可为1:(0.05~1.5),较佳地为1:(0.1~1),例如1:0.43或者1:1。 所述无规共聚物中,较佳地,所述Ar为 较佳地,所述无规共聚物的数均分子量Mn为800~1700,例如1360、1570或者1650。 本发明中,所述硅炔杂化树脂的多分散指数(PDI)较佳地为1~2,更佳地为1.2~ 1.92,例如1.3、1.5、1.59、1.79或者1.91。 本发明中,所述硅炔杂化树脂的硅含量高于24%,较佳地为24~30%,例如 24.8%、26.3%或者27.8%。 一优选实施例中,所述硅炔杂化树脂的数均分子量为1360,n约为12,R1或者R2为甲 基,Ar为 多分散指数为1.59,硅含量为24.8%。 一优选实施例中,所述硅炔杂化树脂为无规共聚物,由通式A和通式B表示的结构 单元组成,通式A和通式B的摩尔比为1:(0 .3~0 .5),数均分子量为1570,多分散指数为 1.79,硅含量为26.3%;封端基为-Ar′-C≡CH-Ar-C≡CH,所述Ar为 一优选实施例中,所述硅炔杂化树脂无规共聚物,由通式A和通式B表示的结构单 元组成,通式A和通式B的摩尔比为1:(0.8~1.2),数均分子量为1650,多分散指数为1.91, 8 CN 111548497 A 说 明 书 4/16 页 硅含量为27.8%,封端基为-Ar-C≡CH,所述Ar为 本发明中,所述可陶瓷化硅炔杂化树脂的加工窗口较宽,可为80~190℃,例如36 ~104℃或者36~96℃。 本发明还提供了一种所述硅炔杂化树脂的制备方法,其包括下述步骤: (1)乙炔格氏试剂与二卤代硅烷经缩聚反应,得端卤代硅炔杂化树脂; 其中,所述乙炔格氏试剂的结构式为:BrMg-C≡C-MgBr;所述二卤代硅烷的结构式 为: R1或R2独立地为氢或甲基,X1或X2独立地为卤素;所述乙炔格氏试剂与所述 二卤代硅烷的摩尔比为1:(1.1-1.5); (2)在有机溶剂存在的条件下,二乙炔基苯基二溴化镁格氏试剂与所述端卤代硅 炔杂化树脂经缩合反应,即可; 其中,所述二乙炔基苯基二溴化镁格氏试剂的结构式为BrMgC≡C-Ar-C≡CMgBr, 其中,Ar为 所述二乙炔基苯基二溴化镁格氏试剂与所述端卤代硅炔杂化树脂的摩尔比至少 为2:1。 步骤(1)中,化学反应式如下: 由上述反应式可知,所述乙炔格氏试剂与所述二卤代硅烷的摩尔比为n:(n 1)。 步骤(2)中,化学反应式如下: 。由上述反应可知,所述二乙炔基苯基二溴化镁格氏试剂与所述端卤代硅炔杂化 树脂经缩合反应可得到主链上σ-π电子离域的二乙炔基苯封端的硅炔杂化树脂。 步骤(1)中,所述乙炔格氏试剂(乙炔基二溴化镁)可通过本领域常规方法制得,例 如可由乙炔溴化镁与烷基溴化镁反应制得,也可由烷基溴化镁与乙炔反应制得。 较佳地,所述乙炔格氏试剂通过下述步骤制得:a.在惰性气氛下将卤代烃加入镁 粉和有机溶剂的混合物中,反应生成烷基格氏试剂;b.在所述烷基格氏试剂中加入“乙炔基 溴化镁或者乙炔”进行反应生成所述乙炔格氏试剂。 上述步骤a的反应式如下所示:R-Br Mg→R-MgBr。 9 CN 111548497 A 说 明 书 5/16 页 上述步骤b的反应式如下所示: HC≡C-MgBr R-MgBr→BrMg-C≡C-MgBr R-H 或者HC≡CH R-MgBr→BrMg-C≡C-MgBr R-H。 步骤a中,所述惰性气氛可通过本领域常规方法获得的不参与化学反应的惰性气 氛,例如氮气气氛。 步骤a中,所述卤代烃可为本领域常规,例如溴代烷。所述溴代烷烃较佳地为溴代 甲烷、溴乙烷、1-溴丙烷和1-溴丁烷一种或多种,更佳地为溴乙烷。 步骤a中,较佳地,在冰水浴条件下,滴加所述卤代烃。 步骤a中,所述卤代烃较佳地以溶液的形式添加。在卤代烃溶液中,卤代烃与溶剂 的质量体积比较佳地为1g:(1~60)mL,更佳地为1g:(5~55)mL例如1g:8mL。在卤代烃溶液 中,采用的溶剂可为本领域常规,一般为醚类溶剂。所述醚类溶剂较佳地为丙醚、四氢呋喃、 2-甲基四氢呋喃、乙二醇二甲醚和1,4-二氧六环中的一种或多种,更佳地为四氢呋喃。所述 溶剂在使用前一般按照本领域常规方法进行无水处理。 根据本领域常识可知,所述卤代烃溶液一般是通过恒压漏斗缓慢滴加入所述混合 物中,所述卤代烃溶液的滴加时间可为本领域常规,较佳地为10~120min,例如30~ 120min。所述卤代烃溶液在使用前一般按照本领域常规方法进行无水处理。 步骤a中,所述卤代烃和所述镁粉的摩尔比可为本领域常规,较佳地为1:(1.01~ 1.5),更佳地为1:(1.05~1.30),例如1:1.25或者1:1.18。 步骤a中,所述镁粉可为本领域常规用于格氏反应的镁粉,一般采用粒径为100~ 400目镁粉,较佳地为150~300目。 步骤a中,所述有机溶剂可为本领域常规的可用于进行格氏反应的有机溶剂,一般 为醚类溶剂。所述醚类溶剂较佳地为丙醚、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和1, 4-二氧六环中的一种或多种,更佳地为四氢呋喃。所述有机溶剂在使用前一般按照本领域 常规方法进行无水处理。 步骤a中,所述有机溶剂和所述镁粉的体积质量比可为本领域常规,较佳地为(2~ 50)mL:1g,更佳地为(10~45)mL:1g,例如34.7mL:1g。 步骤a中,所述有机溶剂和所述卤代烷的体积质量比可为本领域常规,较佳地为(2 ~60)mL:1g,更佳地为(8~55)mL:1g。 步骤a中,所述反应可为本领域常规的格氏反应的条件。所述反应的温度较佳地为 35~45℃,例如40~45℃。所述反应的时间较佳地为1~3h,例如1.5h。 步骤b中,所述乙炔基溴化镁或者乙炔均可市售获得。 步骤b中,所述“乙炔基溴化镁或者乙炔”与所述卤代烷的摩尔比可为本领域常规, 较佳地为1:(1.02~2.2)。 步骤b中,所述“乙炔基溴化镁或者乙炔”较佳地以溶液的形式添加。在“乙炔基溴 化镁或者乙炔”溶液中,“乙炔基溴化镁或者乙炔”与溶剂的摩尔体积比较佳地为(0.1-5) mmol:1mL,更佳地为(0.25-1)mmol:1mL,例如0.5mmol:1mL。 在“乙炔基溴化镁或者乙炔”溶液中,采用的溶剂可为本领域常规,一般为醚类溶 剂。所述醚类溶剂较佳地为丙醚、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和1,4-二氧六 环中的一种或多种,更佳地为四氢呋喃。所述有机溶剂在使用前一般按照本领域常规方法 10 CN 111548497 A 说 明 书 6/16 页 进行无水处理。 步骤b中,较佳地,在冰水浴冷却时,所述“乙炔基溴化镁或者乙炔”溶液通过滴加 的方式加入所述烷基格氏试剂中。所述“乙炔基溴化镁或者乙炔”溶液的滴加时间较佳地为 0.5~2h,例如1h。 步骤b中,所述反应的操作和条件可为本领域常规。所述反应的温度一般为加热回 流。所述反应的时间较佳地为1~3h,更佳地为2h。 步骤(1)中,较佳地X1或X2独立地为氯或者溴。 步骤(1)中,所述二卤代硅烷可为二甲基二卤代甲硅烷和/或甲基氢二卤代甲硅 烷,更佳地为二甲基二氯甲硅烷和/或甲基氢二氯甲硅烷。 其中,当所述二卤代硅烷为二甲基二卤代甲硅烷和甲基氢二卤代甲硅烷的混合物 时,所述二甲基二卤代甲硅烷与所述甲基氢二卤代甲硅烷的摩尔比可为1:(0.05~1.5),较 佳地为1:(0.1~1),例如1:0.43或者1:1。 步骤(1)中,较佳地,将所述二卤代硅烷滴加至所述乙炔格氏试剂中。 其中,所述二卤代硅烷较佳地以溶液的形式添加。所述溶液中,采用的溶剂可为本 领域常规能够溶解二卤代硅烷的溶剂,较佳地为醚类,更佳地为丙醚、四氢呋喃、2-甲基四 氢呋喃、乙二醇二甲醚和1,4-二氧六环,最佳地为四氢呋喃。所述溶剂在使用前一般按照本 领域常规方法进行无水处理。 当所述二卤代硅烷以溶液的形式添加时,所述二卤代硅烷和溶剂的质量体积比可 为(0.12~0.18)g:1mL,例如0.14g:1mL、0.15g:1mL或者0.16g:1mL。 其中,所述滴加的操作和条件可为本领域常规,一般在不断搅拌下滴加所述二卤 代硅烷。较佳地,在冰水浴冷却下进行。所述滴加的速度可为本领域常规,一般在0.5~2.0h 内滴加完毕。 步骤(1)中,所述缩聚反应的条件可为本领域常规,一般为加热回流。所述缩聚反 应的时间较佳地为2-4h。 步骤(1)中,所述缩聚反应结束后,较佳地冷却至室温。 步骤(1)中,所述乙炔格氏试剂与所述二卤代硅烷的摩尔比较佳地为1:(1 .1~ 1.4),更佳地为1:(1.1~1.35),例如6:7、5:6、4:5或者3:4。 步骤(2)中,所述二乙炔基苯基二溴化镁格氏试剂可通过本领域常规方法进行制 备,较佳地通过下述步骤进行: i.在惰性气氛下,将卤代烃加入镁粉和有机溶剂的混合物中,反应生成烷基格氏 试剂;ii.在所述烷基格氏试剂中加入二乙炔基苯进行反应即可;其中,所述二乙炔基苯的 结构式为HC≡C-Ar-C≡CH,其中,Ar为 上述步骤i、ii的反应式如下所示: R-Br Mg→R-MgBr (i) R-MgBr HC≡C-ArC≡CH→BrMgC≡C-Ar-C≡CMgBr R-H (ii) 步骤i中,所述惰性气氛可通过本领域常规方法获得的不参与化学反应的惰性气 氛,例如氮气气氛。 步骤i中,所述卤代烃可为本领域常规,例如溴代烷。所述溴代烷烃较佳地为溴代 11 CN 111548497 A 说 明 书 7/16 页 甲烷、溴乙烷、1-溴丙烷和1-溴丁烷一种或多种,更佳地为溴乙烷。 步骤i中,较佳地,在冰水浴条件下,滴加所述卤代烃。 步骤i中,所述卤代烃较佳地以溶液的形式添加。在卤代烃溶液中,卤代烃与溶剂 的质量比较佳地为1g:2~50g,更佳地为1g:8~45g,例如1g:8g。在卤代烃溶液中,采用的溶 剂可为本领域常规,一般为醚类溶剂。所述醚类溶剂较佳地为丙醚、四氢呋喃、2-甲基四氢 呋喃、乙二醇二甲醚和1,4-二氧六环中的一种或多种,更佳地为四氢呋喃。所述溶剂在使用 前一般按照本领域常规方法进行无水处理。 步骤i中,根据本领域常识可知,所述卤代烃一般是通过恒压漏斗缓慢滴加入所述 混合物中,所述卤代烃的滴加时间可为本领域常规,较佳地为10~120min,例如30~ 120min。所述卤代烃在使用前一般按照本领域常规方法进行无水处理。 步骤i中,所述卤代烃和所述镁粉的摩尔比可为本领域常规,较佳地为1:(1.01~ 1.5),更佳地为1:(1.05~1.30),例如1:1.25。 步骤i中,所述镁粉可为本领域常规用于格氏反应的镁粉,一般采用粒径为100~ 400目镁粉,较佳地为150~300目。 步骤i中,所述有机溶剂可为本领域常规的可用于进行格氏反应的有机溶剂,一般 为醚类溶剂。所述醚类溶剂较佳地为丙醚、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和1, 4-二氧六环中的一种或多种,更佳地为四氢呋喃。所述有机溶剂在使用前一般按照本领域 常规方法进行无水处理。 步骤i中,所述有机溶剂和所述镁粉的体积质量比可为本领域常规,较佳地为(2~ 50)mL:1g,更佳地为(10~45)mL:1g。 步骤i中,所述有机溶剂和所述卤代烷的体积质量比可为本领域常规,较佳地为(2 ~60)mL:1g,更佳地为(10~55)mL:1g,例如10.4mL:1g。 步骤i中,所述反应可为本领域常规的格氏反应的条件。所述反应的温度较佳地为 35~45℃,例如40~45℃。所述反应的时间较佳地为1~3h,例如1.5h。 步骤ii中,所述二乙炔基苯与所述卤代烷的摩尔比可为本领域常规,较佳地为1: (2.1~2.5),例如1:2.2。 步骤ii中,所述二乙炔基苯较佳地以溶液的形式添加。在二乙炔基苯溶液中,二乙 炔基苯与溶剂的质量体积比为较佳地为1g:15mL~1g:25mL,例如1g:19.8mL。 在二乙炔基苯溶液中,采用的溶剂可为本领域常规,一般为醚类溶剂。所述醚类溶 剂较佳地为丙醚、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和1,4-二氧六环中的一种或多 种,更佳地为四氢呋喃。所述有机溶剂在使用前一般按照本领域常规方法进行无水处理。 步骤ii中,较佳地,在冰水浴冷却时,所述二乙炔基苯通过滴加的方式加入所述烷 基格氏试剂中。所述二乙炔基苯的滴加时间较佳地为0.5~2h,例如1h。 步骤ii中,所述反应的操作和条件可为本领域常规。所述反应的温度一般为加热 回流。所述反应的时间较佳地为1~3h,更佳地为2h。 步骤(2)中,较佳地,将所述端卤代硅炔杂化树脂滴加至所述二乙炔基苯基二溴化 镁格氏试剂中。 其中,所述端卤代硅炔杂化树脂较佳地以溶液的形式添加。所述溶液中,采用的溶 剂可为本领域常规能够溶解端卤代硅炔杂化树脂的溶剂,较佳地为醚类,更佳地为丙醚、四 12 CN 111548497 A 说 明 书 8/16 页 氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和1,4-二氧六环中的一种或多种,最佳地为四氢呋 喃。 当所述端卤代硅炔杂化树脂以溶液的形式添加时,所述端卤代硅炔杂化树脂和溶 剂的质量体积比可为0.02~0.03g/mL,例如0.025g/mL。 其中,所述滴加的操作和条件可为本领域常规,一般在不断搅拌下滴加所述端卤 代硅炔杂化树脂。较佳地,在冰水浴冷却下进行滴加。所述滴加的速度可为本领域常规,一 般在0.5~2.0h(例如1h)内滴加完毕。 步骤(2)中,所述二乙炔基苯基二溴化镁格氏试剂与所述端卤代硅炔杂化树脂的 摩尔比较佳地为(2~2.5):1,更佳地为(2~2.2):1。 步骤(2)中,所述缩合反应的操作和条件可为本领域常规,一般为加热回流。所述 缩合反应的时间较佳地为2~4h。 步骤(2)中,所述缩合反应结束后,较佳地进行后处理。所述后处理可为本领域常 规的将端乙炔格氏试剂还原为端炔氢的操作,较佳地按下述步骤进行:将所述缩合反应获 得的反应液与质子试剂混合反应后,水洗、干燥除水、过滤后所得滤液去除溶剂即可。 其中,所述反应液与质子试剂进行混合前,较佳地进行冷却。所述冷却后的温度可 为室温。所述冷却的操作和条件可为本领域常规自然冷却。 其中,所述质子试剂一般以溶液的形式进行添加。当质子试剂以溶液的形式添加 时,采用的溶剂的种类可为本领域常规,较佳地为醚类和/或水。所述醚类可为乙醚。 其中,所述质子试剂的种类可为本领域常规,较佳地为盐酸、氯化铵和冰乙酸中的 一种或多种。 当所述质子试剂以水溶液的形式添加时(例如盐酸水溶液或者氯化铵水溶液),质 子试剂水溶液中,质子试剂的质量分数可为1%~30%,例如5%。当质子试剂以有机溶液的 形式添加时(例如冰醋酸的醚类溶液),质子试剂与有机溶剂的质量体积比可为1g:(60~ 100)mL,例如1g:83mL。 其中,所述质子试剂一般滴加至所述反应液中。 其中,所述混合反应的操作和条件可为本领域常规,一般为搅拌反应1.5-2.5h,例 如2h。 其中,所述水洗的操作和条件可为本领域常规,较佳地水洗至分离出的水相为中 性。所述水洗的操作较佳地按下述步骤进行:将混合反应后所得产物与水(水的用量可与油 相的体积相同)混合均匀后,静置,分离出油相与水相,用水反复洗涤产物,至分离出的水相 为中性即可。 其中,所述干燥除水的操作和条件可为本领域常规,一般用除水剂干燥,静置过 夜。所述除水剂的种类可为本领域常规,例如无水硫酸钠。所述除水剂的用量可为本领域常 规,一般为10%,上述百分比为除水剂与油相的质量百分比。 其中,所述过滤的操作和条件可为本领域常规,一般用过滤漏斗除去除水剂。 其中,去除溶剂的操作和条件可为本领域常规,一般为减压蒸馏即可。所述减压蒸 馏的温度一般比含质子试剂溶液中的溶剂的沸点温度低10℃~20℃。所述减压蒸馏的真空 度可为-0.07MPa~-0.09MPa。 若无特殊说明,步骤(1)(2)均在干燥的惰性气体氛围下进行。所述惰性气体不局 13 CN 111548497 A 说 明 书 9/16 页 限于氦气、氩气等气体,还可包括氮气。 若无特殊说明,步骤(1)(2)均在无水条件下下进行。所述无水条件是指体系中的 水含量在500ppm以下。 本发明还提供了一种硅炔杂化树脂固化物的制备方法,其包括下述步骤:将所述 可陶瓷化硅炔杂化树脂进行固化成型,即可。 本发明中,所述固化成型的操作和条件可为本领域常规。 本发明中,所述固化成型的温度较佳地为150~300℃,例如160℃、170℃、180℃、 200℃、210℃、220℃、240℃、250℃或者260℃。 本发明中,所述固化成型的时间较佳地为2~12h,例如2h、4h、6h或者8h。 本发明中,所述固化成型的操作较佳地为阶梯状升温固化过程,更佳地按下述步 骤进行:首先在160~180℃,保温1.5~2.5h,再升温至200~220℃,保温1.5~2.5h,再升温 至240~260℃,保温3.5~4.5h;更佳地按下述步骤进行:在170℃保温2h,再升温至210℃保 温2h,再升温至250℃保温4h。 本发明还提供了一种如前所述的制备方法制得的硅炔杂化树脂固化物。 本发明中,硅炔杂化树脂复合材料可通过本领域常规复合材料的方法制得,例如 模压成型法。 本发明还提供了一种硅炔杂化树脂复合材料的制备方法,其包括下述步骤: (1)将含有所述可陶瓷化硅炔杂化树脂的溶液浸渍增强纤维后,干燥得预浸料; (2)所述预浸料经热模压工艺即得所述可陶瓷化硅炔杂化树脂复合材料。 步骤(1)中,所述可陶瓷化硅炔杂化树脂的溶液中,所述可陶瓷化硅炔杂化树脂溶 液中树脂的质量分数可为本领域常规,较佳地为30~40%,例如35%。所述溶液中采用的溶 剂可为本领域常规能够溶解所述可陶瓷化硅炔杂化树脂的非质子溶剂,一般可为四氢呋喃 和/或丙酮。 步骤(1)中,所述增强纤维可为本领域常规用于制备复合材料的增强纤维,一般为 碳纤维或者石英纤维。所述碳纤维可为本领域常见编织方式的碳纤维平纹布。所述碳纤维 的种类可为T300碳纤维、T700碳纤维、T800碳纤维或者T1000碳纤维。例如在一优选实施例 中,采用的T300碳纤维平纹布。 步骤(1)中,所述增强纤维的层数可为2~15层,较佳地为10~15层,例如12层。 步骤(1)中,所述浸渍的操作和条件可为本领域常规,一般是在搅拌的条件下,将 所述增强纤维浸渍于所述混合溶液中即可。根据本领域常识可知,一般浸渍完之后,还剩余 少量的溶液(浸渍液),后续将剩余的溶液再涂覆在所述增强纤维或者预浸料的表面,或者 将剩余的少量的溶液(浸渍液)在热模压时,分别均匀涂覆于增强纤维接触的铝箔一侧。 步骤(1)中,所述浸渍的操作之后,一般进行晾干。其中,所述晾干一般是在室温下 自然晾干。 步骤(1)中,所述干燥一般是指真空干燥。所述真空干燥的条件可为本领域常规。 所述真空干燥的温度较佳地为50~70℃,例如60℃。 所述真空干燥的真空度较佳地为-0.07MPa以下,例如-0.09MPa。 所述真空干燥的时间较佳地为1~4h,例如2~3h。 所述干燥的终点较佳地为干燥至预浸料中挥发分质量分数小于10%。 14 CN 111548497 A 说 明 书 10/16 页 步骤(2)中,所述热模压工艺为本领域常规的模压固化成型工艺,其操作和条件可 为本领域常规,一般在平板硫化机上进行。根据常识可知,一般将所述预浸料夹于两层铝箔 之间后,放于两平板或模具中,移至平板硫化机,进行热模压成型即可。其中,所述平板或模 具的材质可为本领域常规的钢制平板或模具。 步骤(2)中,所述热模压工艺中,温度较佳地为150~300℃,例如160℃、170℃、180 ℃、200℃、210℃、220℃、240℃、250℃或者260℃。 步骤(2)中,所述热模压工艺中,模压时间较佳地为2~12h,例如2h、4h、6h或者8h。 步骤(2)中,当热模压工艺中采用的是平板时,压力较佳地为2~3MPa,例如2MPa。 步骤(2)中,所述热模压工艺的操作较佳地为阶梯状升温固化过程,更佳地按下述 步骤进行:首先在160-180℃,保温1.5~2.5h,再升温至200~220℃,保温1.5~2.5h,再升 温至240~260℃,保温3.5~4.5h;更佳地按下述步骤进行:在170℃保温2h,再升温至210℃ 保温2h,再升温至250℃保温4h。 本发明中,根据常识可知,所述热模压工艺制得的复合材料形态为板材。 本发明还提供了一种由上述制备方法制得的硅炔杂化树脂复合材料。 本发明还提供了一种陶瓷材料的制备方法,其包括下述步骤:将所述硅炔杂化树 脂固化物或者所述硅炔杂化树脂复合材料进行热解陶瓷化即可。 本发明中,所述热解陶瓷化的操作和条件可为本领域常规。所述热解陶瓷化的温 度较佳地为200~1500℃,例如300℃、500℃、1000℃或者1450℃。所述热解陶瓷化时的压力 一般为常压。 本发明中,所述热解陶瓷化较佳地按下述步骤进行:首先升温至250~350℃,然后 升温至450~550℃,再升温至1400~1500℃保温,再降温至450~550℃。 更佳地按下述步骤进行:首先升温至250~350℃,然后升温至450~550℃,再升温 至1400~1500℃保温4~8h(例如6h),再降温至450~550℃后自然冷却。所述烧结结束后, 一般自然冷却即可。 在一较佳地实施例中,按下述步骤进行:按照5℃/min升至300℃,2℃/min升至500 ℃,1℃/min升至1450℃并保持6h,再以2℃/min降至500℃后自然冷却的程序进行烧结。 本发明中,所述热解陶瓷化的过程中,升温或降温的速率可为本领域常规,较佳地 为0.5~10℃/min,例如1℃/min、2℃/min或者5℃/min。 本发明还提供了一种由上述制备方法制得的陶瓷材料。 在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实 例。 本发明所用试剂和原料均市售可得。 本发明的积极进步效果在于: (1)本发明通过在高硅含量的硅乙炔树脂链端引入二乙炔基苯,制备得到具有主 链的σ-π电子离域的共轭结构的可陶瓷化硅炔杂化树脂。本发明的硅炔杂化树脂可易溶解 于丙酮、THF、乙腈、乙酸乙酯、乙醚、氯代烃溶剂和DMF、DMSO等强极性溶剂中,具有良好的可 加工特性(加工窗口大于100℃)和优良的耐热性。 (2)本发明的硅炔杂化树脂在加热下发生固化反应(固化反应温度较低,初始固化 温度低于200℃,例如可为150℃),可满足热模压、树脂转移模塑和缠绕成型工艺。固化交联 15 CN 111548497 A 说 明 书 11/16 页 提高聚合物的分子量,固化时无小分子放出,所得固化物具有优异的热性能和良好的力学 性能。 本发明的硅炔杂化树脂硅含量高于24%,在高温下可形成陶瓷材料,陶瓷化率高 (裂解时保留更多的Si和C(形成的碳化硅中Si的质量分数高于20%);本发明的硅炔杂化树 脂在航空航天领域的应用前景好。 (3)本发明制得的碳纤维增强硅炔杂化树脂复合材料,硅炔杂化树脂与碳纤维增 强体间有良好的浸润性,其复合材料力学性能良好,例如弯曲强度大于240MPa,弯曲模量高 于40GPa,层间剪切强度高于12MPa,可作为基体树脂和陶瓷前驱体应用。 附图说明 图1为实施例1制得的硅炔杂化树脂的FT-IR谱图。 图2为实施例1制得的硅炔杂化树脂的1H-NMR谱图。 图3为实施例1制得的硅炔杂化树脂的流变曲线。 图4为实施例1制得的硅炔杂化树脂的DSC曲线图。 图5为实施例1制得的硅炔杂化树脂固化物在氮气条件下的TGA曲线。 图6为实施例1制得的硅炔杂化树脂的固化物烧结后所得产物的XRD图谱。











