
技术摘要:
本发明公开了一种提高亲水性药物包封率的微球制备方法,包括以下步骤:步骤(a):制备含有亲水性药物的内水相及含有聚合物载体的油相;步骤(b):混合内水相和油相形成初乳液,初乳液形成后快速降温以增加初乳粘度至1000~2000cp;步骤(c):将步骤(b)中粘度增加后的初乳 全部
背景技术:
与传统制剂相比,长效缓释制剂可以减少给药次数,提高患者顺应性,降低副作 用,提高疗效,在临床上已凸显优势,近年来已成为药物新剂型研发的热点。截止目前,国外 共有10余个微球长效品种上市,其中除了利培酮、纳曲酮,盐酸米诺环素外,其余均为亲水 性药物分子,包括多肽、蛋白类药物。 多肽蛋白类药物因具有效价高、生物相容性好、剂量低等特征,在疾病的预防、诊 断和治疗中担任越来越重要的角色。但这类亲水性药物通常体内半衰期短,为达到药物疗 效需频繁给药,导致患者顺应性差。为提高多肽蛋白类药物生物利用度,降低给药频率,其 长效剂型被开发以满足临床需求。 微球是指药物分散或被吸附在高分子、聚合物基质中而形成的微粒分散体系,借 由微球表面载体的缓释性能,可实现微球在体内的长效给药,是目前长效制剂的主要制剂 形式。制备微球的载体材料很多,主要分为天然高分子和合成聚合物。聚乳酸(Poly(lactic acid),以下简称PLA)和聚乳酸乙醇酸共聚物(Poly(lactic-co-glycolic acid),以下简称 PLGA)是迄今研究最多、安全性最好、应用最广的合成聚合物载体材料。 复乳法最早被用于包封多肽类药物,随着技术的不断进步,目前已被成功应用于 多肽类药物微球的开发,例如Lupron Depot(醋酸亮丙瑞林)。该方法首先将水溶性多肽蛋 白类药物溶于内水相,聚合物载体溶于有机相,内水相加入有机相乳化形成初乳,然后再将 初乳转移至含乳化剂的外水相中形成复乳,之后缓慢搅拌,并采用气体吹扫或减压蒸发方 式去除有机溶剂,固化形成微球。但复乳法由于技术本身的限制,所生产的微球包封效率普 遍偏低,这导致了昂贵的多肽原料药的浪费,显著增加研发及生产成本。 公开号为US5631020A的专利文献中记载了基于内水相(W1)/油相(O)/外水相(W2) 形成的高分子微球的控释制剂中,通过向内水相中添加赋形剂将其粘度提高至5000cp以 上,以增加药物扩散阻力来降低液中干燥过程中药物向外水相的渗漏,并由此提高药物包 封效率。此类赋形剂已被应用于上市产品中,如武田醋酸亮丙瑞林产品中为了提高包封效 率在内水相中添加了明胶,但明胶存在致敏性问题已成公知。 公开号为US4954298A专利文献中记载了基于内水相(W1)/油相(O)/外水相(W2)形 成的高分子微球的控释制剂中,通过降低初乳温度至特定范围内在一定程度上能够增加初 乳粘度,使初乳在外水相中分散后药物扩散的传质阻力增加,并由此提高药物包封效率。 可见,在W1/O/W2型含亲水性药物的聚合物微球的开发过程中,已发展出一些特殊 甚至极端的工艺来提高药物包封效率来降低生产成本,例如提高油相浓度、内水相浓度、制 备时通过控制低温以获得较高的初乳粘度,来抑制药物向外水相的渗漏。这些方式固然是 有效的,但过高的油相浓度、内水相浓度又会造成工业生产中过滤除菌困难,过高的初乳粘 3 CN 111568877 A 说 明 书 2/12 页 度会造成转移损失增加、牺牲产率,无疑给微球产业化带来新的重重困难。 发明人在长期的产品开发工作中发现,提高初乳粘度至2000cp左右时,包封率可 以从60~70%(常规复乳法)提高至85~90%,但,但在干燥过程中存在较为严重的药物损 失现象;当继续提高初乳粘度至2000~5000cp甚至更高时,此时包封率会进一步提高至90 ~95%水平,但在这个高粘度状态下初乳分散过程受阻,在复乳液滴未剪切形成圆整球体 之前,表面聚合物材料便析出、固化成形,往往会造成微球粒度不均一,形态不圆整,呈现丝 状、橄榄状、蝌蚪状等多样形态共存,降低临床给药通针性,增加注射困难性。这些具有不规 则形状的微球在离心收集过程中,也会形成架桥作用,导致固液分离不彻底,微球沉淀不紧 实,上清液浑浊,在上清液去除的过程中会带走大量的微球,造成收率下降。 因此,基于当前现有技术,为了提高微球药物包封效率,降低生产成本,有必要研 究新的思路来提高包封效率。
技术实现要素:
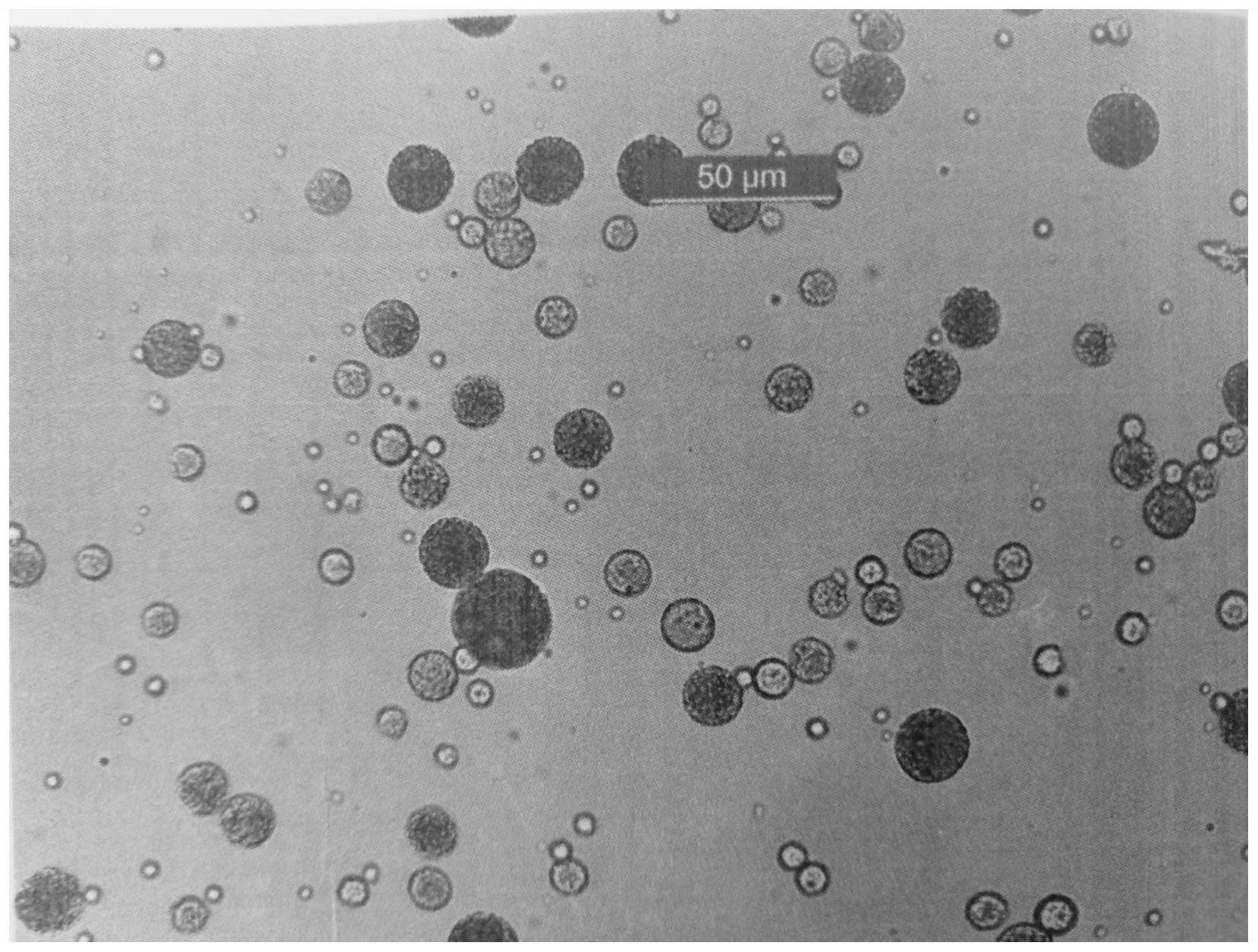
本发明的目的在于提供一种提高亲水性药物包封率的微球制备方法,该方法可显 著提高药物包封率,并且可制备具有高载药量、包封率接近100%的亲水性药物微球。 本发明解决其技术问题所采用的技术方案是: 一种提高亲水性药物包封率的微球制备方法,包括以下步骤: 步骤(a):制备含有亲水性药物的内水相及含有聚合物载体的油相; 步骤(b):混合内水相和油相形成初乳液,初乳液形成后快速降温以增加初乳粘度至 1000~2000cp;采用快速降温方法获得的初乳粘度在1000~2000cp,复乳形成过程中不存 在分散不彻底、微球黏连、形态不圆整或拉丝严重等现象; 步骤(c):将步骤(b)中粘度增加后的初乳液以体积比1:100~1:200混合分散于外水相 中,获得复乳液,形成的复乳液立即保持静止5~10min,得到初产物; 步骤(d):将上述初产物进行第一轮干燥获得湿微球,所述湿微球的有机溶剂残留量不 高于0.5%(w/w),之后进行第二轮干燥去除水分及残留溶剂后获得微球终产物。 发明人在进行复乳法制备亲水性药物微球产品的研发开发过程中,发现了影响药 物包封效率的关键环节。该关键环节主要集中于复乳制备过程及对微球的干燥过程,即通 过特定控制复乳制备工艺及液中干燥的参数,可制备具有高载药量、包封率接近100%的亲 水性药物微球。 本发明发现的影响药物包封效率的关键环节之一为:复乳液形成后应立即保持静 止5~10min。当静止时间低于5min时,会存在复乳液外壳层固化不完全的现象,导致后续液 中干燥过程中液体的轻微搅动也会造成药物的大量泄露;静止时间高于10min时,复乳液之 间会形成架桥作用,有机溶剂不能及时去除,微球沉降、聚集在反应罐底部,出现片状或块 状聚集体,使成品微球粒度增加(该步骤所述的影响包封率的关键参数在本发明中第一次 公开)。优选的,复乳液形成后应立即保持静止6~8min。 在本发明保护范围内,复乳液保持静止一段特定时间内,并不会造成微球的聚集 和粘连,这得益于微球制备过程中的初乳粘度的控制、初乳有机溶剂含量的控制及初乳与 外水相之间的体积比,使得复乳液形成后表面迅速固化,形成薄的外壳层,使球体之间保持 独立,即使在不搅动的前提下也不会发生聚集和粘连。 4 CN 111568877 A 说 明 书 3/12 页 步骤(d)中,所述湿微球的有机溶剂残留量不高于0.02%。 步骤(d)中,第一轮干燥采用循环式液体交换方式,在15~25℃温度条件下进行液 中干燥2.5~5h并辅以气体吹扫。 本发明发现的影响药物包封效率的关键环节之二为:将静止后的复乳液保持15~ 25℃的温度条件下,进行液中干燥,实现方式为进行循环式液中干燥并辅以气体吹扫。采用 循环式液体交换方式进行液中干燥是为了降低液体扰动程度,防止微球在机械力的作用下 向外水相渗漏,辅以气体吹扫的目的是增加有机溶剂去除速率,加快微球固化进程。 所述循环式液体交换方式为通过将反应罐底部液体不间断的返回至反应罐中液 面上方,液体循环频率为0.5-2次/min,循环频率计算公式为单位时间内液体流量除以反应 罐内液体总量。用于将反应罐底部液体不间断的返回至反应罐液面上方的输送设备可以是 泵,优选地,可以为离心泵、往复泵、旋转泵、转子泵,更优选地,可以为离心泵。反应罐内液 体循环效果应满足在单位时间内液体循环频率应为0.5~2次/分钟,循环频率计算公式为 单位时间内液体流量(单位:L/min)除以反应罐内液体总量(单位:L)。 所述液中干燥过程气体吹扫出口流量不低于100L/min,且保证反应罐上方气体更 换频率不低于5次/分钟。 液中干燥过程气体吹扫高度位于反应罐内液面上方不超过10cm的高度范围内。 显而易见,采用上述循环式液中干燥方式并辅以气体吹扫既可以使外水相扰动程 度降至最低,还可以满足快速的有机溶剂去除速率,二者相辅相成进一步降低了微球固化 过程中的药物泄露程度。需要说明的是,本发明所述的提高药物包封率的最重要方式仍为 复乳液形成后的静止保持阶段,该阶段为复乳法制备微球药物损失最关键的环节,影响程 度高于液中干燥过程。 优选地,所述液中干燥过程持续通气时间应不低于2.5h且不高于5h,液中干燥终 点应保证湿微球内溶剂残留水平不得高于0.5%(w/w)。当保持时间小于2.5h时,溶剂去除 不彻底,离心过程中微球之间产生黏连和聚集,不利于冻干前湿微球的分散过程;当保持时 间高于5h时,药物包封效率会进一步下降,长时间与外水相接触会使微球内药物分布改变、 向外壳层靠近,造成突释效应。 由于液中干燥过程对微球的包封过程(包封率)及形成的药物分布状态(释放度) 极其重要,尤其是液体循环频率的影响最为显著,在本发明的实施例和对比例中,详细的列 举了不同的液中干燥参数对微球关键质量属性的影响,表征指标主要包括:成品微球的粒 度、药物包封率及载药量。 采用本发明所述的液中干燥工艺参数时,微球药物包封率接近100%,释放过程中 不存在突释及后期释放不完全的现象。 作为优选,当液中干燥待去除的溶剂为二氯甲烷时,液中干燥时间为3h-4h,保证 液中干燥终点湿微球内溶剂残留水平不高于0.02%。 步骤(b)中,初乳液形成后快速降温至15~18℃。 步骤(c)中,外水相的温度为15~18℃。 步骤(b)中,内水相和油相的重量比为1:5-20。优选地,内水相和油相的重量比为 1:10-15。 所述第二轮干燥的方法为流化床干燥、恒温浴干燥、真空减压干燥或冷冻干燥中 5 CN 111568877 A 说 明 书 4/12 页 的任意一种。 作为优选,第二轮干燥的方法为冷冻干燥时,主干燥温度为-10~-5℃,保持时间 15~20h,后期解析干燥温度范围为45~55℃,后期解析干燥时间不低于24h。冷冻干燥之 前,可以通过加入冻干保护剂来提高微球稳定性、降低冻干过程中微球的聚集。优选地,可 向上述洗涤、浓缩后的湿微球混悬液中加入冻干保护剂(例如,糖类),随后进行冷冻干燥, 更优选地,冻干保护剂可以为甘露醇。冷冻干燥目的为去除微球中残余水分及有机溶剂,进 行冷冻干燥的条件为:主干燥温度为-10~-5℃,保持时间15~20h,后期解析干燥温度范围 是45~55℃,优选地,保持后期解析干燥时间应不低于24h,更优选地,后期解析干燥保持时 间应不低于48h。 本发明的亲水性药物包含亲水性化合物、肽、蛋白质、抗体、核酸或它们的盐。优选 地,本发明的亲水性药物可以为具有生理学活性的多肽,更优选地,可以为醋酸亮丙瑞林。 亲水性药物与水性溶剂混合形成内水相,本发明所述的水性溶剂可以为水、低级 醇、乙腈、丙酮、四氢呋喃中的一种或几种的混合物,优选地,本发明的水性溶剂为水。 形成内水相(W1)和药物的重量比可根据水性溶剂的种类、药物的种类而不同,但 不限定于此,例如以水性溶剂:药物=1:1.2~1:0.2的重量比混合,优选地,以水性溶剂:药 物=1:1.2~1:0.8的重量比混合,更优选地,相对于1重量份的水性溶剂,药物为1重量份的 比率进行混合。 可以以药物加入水性溶剂或水性溶剂加入药物的方式在加热条件下混合内水相, 优选地,加热条件为40~80℃,更优选地,55~65℃。 所述聚合物载体是指形成微球外壁的高分子聚合物,本发明所述的聚合物载体是 指具有脂肪碳链的聚酯类高分子,可以为聚乳酸、聚乳酸羟基乙酸共聚物(乙交酯丙交酯共 聚物)、聚已内酯、聚羟基丁酸酯、聚亚胺基碳酸酯、聚酸酐、聚氨基酸、乳酸和氨基酸共聚 物,优选地,可以为聚乳酸和聚乳酸羟基乙酸共聚物,更优选地,可以为聚乳酸羟基乙酸共 聚物。在本发明的聚合物微球的制备方法中使用的聚合物载体重均分子量未特别限制,但 通常可以为5,000~130,000,更优选地,可以为10,000~20,000。 所述聚乳酸和聚羟基乙酸共聚物(PLGA)的乳酸和乙醇酸的摩尔比可以为0:100~ 100:0,优选地,可以为50:50~85:15(50:50、75:25、85:15),更优选地,可以为75:25。 油相采用脂溶性溶剂溶解聚合物载体,脂溶性溶剂可以为二氯甲烷、三氯甲烷、乙 酸乙酯、乙酸丁酯,优选地,可以为二氯甲烷和三氯甲烷,更优选地,可以为二氯甲烷。 在能够溶解聚合物载体的前提下,对脂溶性溶剂的量不作限定,脂溶性溶剂的量 可根据聚合物的种类和脂溶性溶剂的种类而不同,可以为聚合物载体重量的1~10倍,优选 的,可以为1~5倍,更优选的,可以为聚合物载体重量的1~2倍。 油相(O)可通过在脂溶性溶剂中混合聚合物的方法制备,溶解温度通常为室温范 围(25±10℃),根据需要可选择振荡混合或静置溶解。 初乳液通过混合内水相和油相的方法制备,所述混合可通过已知的方法进行,可 以为均质器法、超声波法、旋浆式搅拌器法、涡轮式搅拌器法,优选地,可以为均质器法。优 选地,本发明的W1/O初乳液可通过在油相中加入内水相,并通过均质器进行均质化的方法 制备。 油相和外水相的混合比应介于1:100~1:200,具体地,相对于1体积份的油相,能 6 CN 111568877 A 说 明 书 5/12 页 够以100~200体积份的比率混合外水相,更优选地,相对于1体积份的油相,能够以100~ 150体积份的比率混合外水相。当外水相高于200体积份时,会造成球形不圆整;当外水相低 于100体积份时,会发生药物泄漏。 本发明中收集微球的方法为常规的固液分离方法,优选地,可以为离心分离、过滤 收集,更优选地,通过连续流离心机进行离心收集、洗涤。洗涤步骤是将浓缩的湿微球重新 分散在去离子水中再收集,洗涤的目的是去除湿微球表面吸附的游离药物、乳化剂及有机 溶剂。 当通过本发明的微球制备方法来制备聚合物微球时,可获得具有极低的粒度,极 高载药量、包封率接近100%且球形更圆整的亲水性药物微球。众所周知,微球的载药量与 其平均粒径和比表面积呈反比,通常高的平均粒径意味着高的载药量,即载药量和平均粒 径不能分开而论。而本发明所述的方法制备的微球平均粒度极低,甚至不超过10um,仍旧可 以实现高的药物装载率和包封效率。因此,与现有技术相比,本发明所述的方法在复乳法药 物包封率低的现有技术基础上取得了突破性进展。 与通过现有的方法来制备的聚合物微球相比,本发明的聚合物微球具有接近 100%的药物包封水平,球形更圆整,同时极低的粒度分布以便于临床注射以及可控的药物 释放。 本发明的有益效果是:采用本发明所述的方法制备微球,能够获得同时具有高包 封率、高载药量和极低粒度的多肽微球;这不仅节约了多肽微球的研发及生产成本,同时又 降低病人注射过程中的疼痛性,在达到同等疗效的前提下节约了资源,提高了竞争力。 附图说明 图1是本发明实施例1所制备的微球的扫描电镜图。











